(주)인실리코젠이 함께한 고추 유전체 프로젝트 연구 결과 - 생명과학 분야 최고 학술지 Nature Genetics (IF 35.2)의 1월 19일자 온라인 판에 게재
- Posted at 2014/01/28 22:07
- Filed under 회사소식

관련기사
고추 매운맛 유전자, 국내 연구진이 밝혀냈다 2014.01.22 티브이데일리
고추 유전체서열 국내 독자 기술로 완성 2014.01.21 정책브리핑
고추 유전체서열 국내 독자 기술로 완성 2014.01.20 아시아투데이
고추 표준 유전체 염기서열 국내 기술로 완성 2014.01.20 연합뉴스
좀 더 맵고 맛있는 고추 나온다 2014.01.20 동아사이언스
논문바로보기
http://www.nature.com/ng/journal/vaop/ncurrent/full/ng.2877.html
'고추' 하면 떠오르는 친근감은 비단 우리나라 뿐만이 아닐 것입니다. 고추는 세계적으로 사랑 받고 있고 영양학적인 가치 또한 우수하여 토마토, 감자와 함께 대표적인 작물 중 하나로 꼽히고 있습니다. 그러나 생물학자들에게는 대중적인 선호도 이외에 토마토, 감자와 함께 고추에서 밝히고자 하는 흥미로운 관심 거리가 있습니다. 서로 닮은 듯 아닌 듯 한 이들 세 작물은 모두 가지과 (Solanaceae)에 속하는 것으로 진화와 육종을 통해 얻어진 공통된 특성과 특이적인 특성을 각각 분자적으로 밝히기에 좋은 모델이 되기 때문인데요, 특히 토마토와 고추의 경우 흥미로운 연구거리가 가득합니다.
첫번째, 토마토의 경우 사과나 바나나와 같이 에틸렌 가스에 의해 후숙성이 촉진되는 climateric fruit 인 반면, 고추는 포도와 같이 후숙성이 촉진 되지 않는 non-climateric fruit으로 같은 가지과 작물로써 서로 비슷한 유전자 세트를 가지면서도 서로 다른 형태의 숙성과정을 거치게 되는 메카니즘은 무엇일까?
두번째, 토마토의 유전체는 약 900Mb정도인데 반해 고추는 약 3Gb에 달하는 거대한 유전체 사이즈를 갖는 이유는 무엇일까?
세번째, 고추의 대중적인 인기의 근간이 되는 매운맛 성분인 캡사이신의 생합성 경로는 어찌 될까? 이 런 모든 질문에 대한 해답이 최근 생물정보 컨설팅 전문기업인 (주)인실리코젠에서도 참여한 서울대 최도일 교수님 연구팀에서 Nature genetics 에 발표한 논문 Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species http://www.nature.com/ng/journal/vaop/ncurrent/full/ng.2877.html 에서 모두 해결되었습니다.

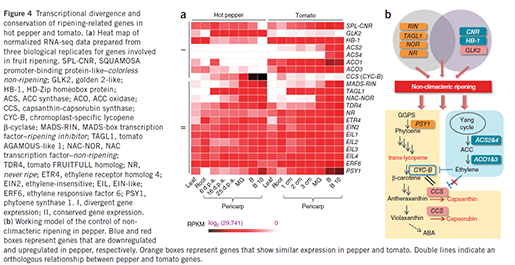
논문에 많은 내용들이 있지만 그 중 후숙성 과실인 토마토와 그렇지 않은 고추와의 과실 숙성 메카니즘의 차이를 보여주는 마지막 메인 figure를 살펴보면, ripening 관련 유전자는 두 종 모두에서 보존되어 있으나 그림에서 보여지는 것과 같이 mRNA상의 발현의 차이로 (group I) 표현형의 차이가 유발된 것으로 나타났습니다. 이 중 주요 유전자는 ethylene이 생성되는 과정에 수반되는 유전자들의 발현이 고추에서 모두 저하되어 ethylene 생성이 저하되고 그로 인해 ethylene에의해 repression되는 CCS(capsanthin-capsorubin synthase)의 발현이 tomato에 비해 월등히 높게 나타나고, 결국 pepper-specific carotenoids인 Capsanthin, capsorubin의 합성이 높아 tomato와는 다른 표현형을 나타냈습니다. 반면, tomato에서는 CCS와 ortholog 관계를 갖는 CYC-B(chromoplast-specific lycopene beta-cyclase) 유전자의 발현이 ripening 과정 동안 ethylene의 높은 합성으로 인해 억제됨을 나타냄으로써 그 메커니즘을 밝혔습니다.
Comparative fruit ripening
이 외에도 고추에 많은 비타민 함량의 메커니즘이라던가, 토마토와 고추의 과실이 물러지는 차이의 원인 메커니즘과 같은 유전체 전문가가 아니더라도 흥미를 가질 만한 많은 내용이 담겨 있습니다. 물론 유전체 전문가(?)의 입장에서도 소중한 정보가 가득합니다. 사실 제가 마지막 figure만을 소개한 이유는 이 하나의 figure를 위해 수행되어야 하는 genome assembly(유전체 서열 완성), gene structure분석(유전자의 서열 및 구조, 유전자 기능, 유전체내 전체 유전자 세트), gene family분석(ortholog, paralog분석) , genome expansion분석( repetitive sequence분석), gene expression 분석(transcription factor분석, RNAseq 분석, pathway 분석), genome variant 분석(SNP, indel 분석), phylogeny 분석과 같은 많은 분석이 수반되어야 하고, 이러한 정보는 supplementary information에서 제공하고 있는 table 54개, figure 49개에 고스란히 담겨져 있음을 알려드리고 싶어서 입니다. 이들 데이터는 마지막 figure와 같은 많은 생물학자들에게 실마리를 제공할 리소스 데이터로 제공이 될 것이기에 그 잠재력이 더욱 큽니다.
Gene structure분석 파이프라인
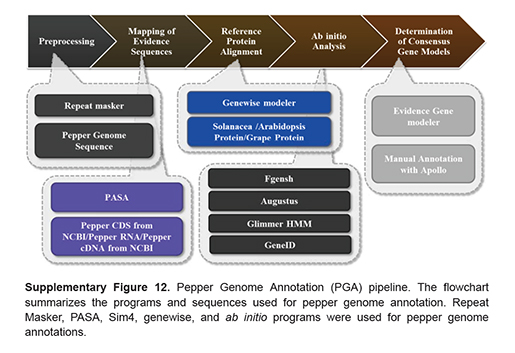
유전자 구조 분석 파이프라인으로 고추 유전체 분석을 위해 고추의 mRNA(RNAseq, ESTs)서열, 단백질 서열, 토마토 및 감자의 단백질 서열, 애기장대, 포도 및 가지과 작물의 단백질 서열을 이용한 Evidence gene modeling과 여러개의 ''ab initio'' gene modeling (gene prediction)이 함께 수행되어 이들의 공통된 유전자 모델을 선정하는 combined gene modeling이 수행되었습니다. - (주)인실리코젠 지원
마지막으로 이번 연구의 가장 큰 성과라면, 순수 국내 연구진의 기술로 이뤄졌다는 점과 생물정보의 학문적 발전입니다. 식물의 유전체에는 유전자 영역 이외에 repeat 영역이 포유류나 균류, 미생물에 비해 매우 많이 존재하기 때문에 실제 유전체 서열을 완성하기에 매우 까다로운 조건을 갖고 있습니다. 단적으로 토마토, 감자의 경우 국제 컨소시엄을 통해 전세계 연구진의 협업에 의해 이뤄진 점만 보더라도 고추 유전체의 완성은 의미가 크다고 할 수 있습니다. 더욱이 유전체 크기가 토마토에 비해 3배이상 커지고 커진 대부분이 repetitive sequence에 해당하는 LTR retrotransposons 임을 감안하면 유전체 서열 어셈블리만 보더라도 많은 노력이 수반됐음을 알 수 있습니다. 실제, 오픈 소스 프로그램(SOAPdenovo, SSPACE, FLAKE)과 상용 프로그램(CLC Assmebly Cell; CLCbio사, 서울대, (주)인실리코젠의 공식 MOU를 통한 지원)이 모두 이용되었으며, 시퀀싱 또한 다양한 플랫폼/디자인으로 여러번의 수정과 시도를 반복하며 현재의 결과를 얻어냈습니다. 뿐만 아니라 유전자 구조 분석 또한 세계적인 수준의 분석이 진행되었으며 genome expansion, gene expression, 진화적론적인 phylogenetic 분석 모두 국내 연구진들의 몰입적인 연구를 수행한 결과라 할수 있습니다.
다시한번, 생물정보 컨설팅을 전문으로 하는 (주)인실리코젠의 입장으로 NGS라는 막강한 도구와 나날이 정신없이 발전하고 있는 생물정보학의 발전을 통해 보다 많은 좋은 소식이 있기를 기대해 봅니다.
Codes사업본부 Research실
선임컨설턴트 신윤희 선임
선임컨설턴트 신윤희 선임
Posted by 人Co
- Tag
- Bioinformatics, Comparative genomics, insilicogen, Nature Genetics, NGS, Phylogeny, SNP, 가지과, 고추 유전체, 생물정보학, 유전체, 인실리코젠, 토마토
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/145