BKL TRANSFAC
- Posted at 2010/04/27 14:55
- Filed under 제품소식
Biobase의 대표적인 제품군인 TRANSFAC은 eukaryotic gene regulation을 분석하기 위한 최적의 기초 데이터를 제공하고 있다. Transcription factors, miRNAs, 그리고 이들과 관련된 유전자의 프로모터 정보를 비롯하여 ChIP-Seq 데이터로부터 1,000,000건 이상의 binding sites 정보, 57,000건 이상의 human RNA polymeraseII의 위치정보를 포함하고 있다. 이들 정보는 모두 실험적으로 증명 되었거나 논문에 게재된 정보를 전문가의 리뷰를 통해 정확하면서도 통합적인 이해를 할 수 있도록 하였다.
2010년 현재 TRANSFAC®의 데이터베이스는 DNA binding, expression 그리고 regulation에 관련한 전문가의 manual curation을 다음과 같이 수행하였다.

이들 데이터는 실험적으로
TRANSFAC® Professional은 공개된 데이터에 비해 약 4년 정도의 데이터가 업데이트되어 있는 상태로 그 데이터양은 promoter서열이 약 280,000건, 700,000건의 ChIP-chip/-Seq 데이터를 더 포함하고 있다(figure 1).
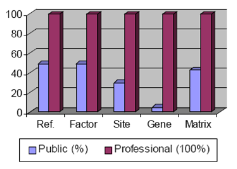 Figure 1. Public database와 Professional version의 데이터양의 차이
Figure 1. Public database와 Professional version의 데이터양의 차이
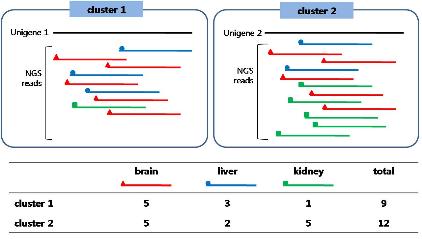
특정한 발현 패턴을 보이는 유전자의 발현 조절 메커니즘을 분석 하고자 할 때 기본적으로 유전자의 upstream 영역에서 작용하는 transcription factor(TF)를 알아보게 된다. TRNASFAC®은 기본적인 transcription factor 및 binding site에 대한 정보를 제공함과 동시에 미지 서열에 binding 가능한 transcription factor를 예측할 수 있는 MatchTM, PatchTM, 그리고 Catch® 프로그램도 제공하고 있다(Figure 4).
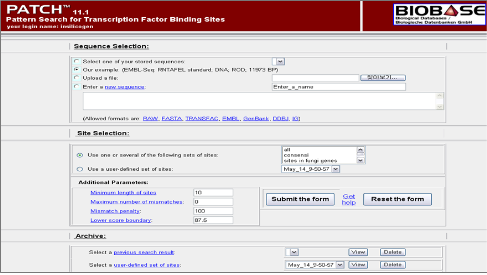 Figure 4. TRANSFAC Professional의 TF search를 위한 PATCH. Pattern match를 통한 미지의 서열에 binding 가능한 TF를 search한다. 이때 false positive를 최소화하기 위해 찾고자 하는 TF의 종 정보를 제한하여 식물 유전자의 경우 식물 데이터베이스를 사용하고 mamalian 유전자의 경우 mamalian 데이터베이스를 사용한다. 또한 특정 찾고자 하는 TF만을 대상으로 할 경우 분석자에 의해 선택된 TF만으로 구성된 프로파일을 제작하여 분석할 수도 있다.
Figure 4. TRANSFAC Professional의 TF search를 위한 PATCH. Pattern match를 통한 미지의 서열에 binding 가능한 TF를 search한다. 이때 false positive를 최소화하기 위해 찾고자 하는 TF의 종 정보를 제한하여 식물 유전자의 경우 식물 데이터베이스를 사용하고 mamalian 유전자의 경우 mamalian 데이터베이스를 사용한다. 또한 특정 찾고자 하는 TF만을 대상으로 할 경우 분석자에 의해 선택된 TF만으로 구성된 프로파일을 제작하여 분석할 수도 있다.
MatchTM는 TF의 binding site를 matrix로 구성하여 찾는 방법이며, PatchTM는 서열의 pattern match 방법을 이용하여 찾는 방법이다. Catch®는 composite elements를 찾고자 할 때 사용하게 되는데 보통 이들 프로그램을 모두 사용하여 가능한 모든 TF를 찾고 실험에 이용한다. 또한 실험적으로 하나하나 규명할 수도 있으나 유전체 전체 유전자를 대상으로 분석하고자 할 때, 웹으로 운영되는 다음 프로그램에 서열을 하나씩 분석하기는 매우 어려우므로 local 서버나 PC에 설치하여 batch로 서열을 분석할 수도 있다. 이후 얻어진 유전자의 upstream 영역에서 작용하는 TF의 profile정보는 통계적 기법을 통해 유의한 TF를 선별하기도 하고, 데이터베이스화하기도 한다.
또한 얼마 전 덴마크의 CLCBio사와의 협력을 통해 CLCMainWorkbench 혹은 CLCGenomicsWorkbench의 plug-in 기능을 통해 TF정보를 visualization 할 수도 있게 되었다. 따라서 NGS에 의한 RNA-seq 정보 및 유전자 발현정보와 함께 전사조절 , 메커니즘까지 확대하여 함께 분석할 수 있는 최적의 데이터를 제공하고 있는 것이다.

2010년 현재 TRANSFAC®의 데이터베이스는 DNA binding, expression 그리고 regulation에 관련한 전문가의 manual curation을 다음과 같이 수행하였다.
이들 데이터는 실험적으로
- transcription factor binding site나 혹은 composite elements를 증명하고자 할 때,
- promoter sequence를 찾고자 할 때
- miRNA targets을 찾고자 할 때
- 관심 있는 영역에 binding 가능한 transcription factor를 찾고자 할 때
- transcription factor들 간의 조절을 알고자 할 때
TRANSFAC®의 데이터 구성
TRANSFAC® Professional은 공개된 데이터에 비해 약 4년 정도의 데이터가 업데이트되어 있는 상태로 그 데이터양은 promoter서열이 약 280,000건, 700,000건의 ChIP-chip/-Seq 데이터를 더 포함하고 있다(figure 1).
이들의 자세한 내용은 figure 2에서 보여 지는 것과 같이 transcription factor의 서열 정보를 비롯한
binding 가능한 site정보, 도메인정보, regulation 정보를 총체적으로 담고 있다.
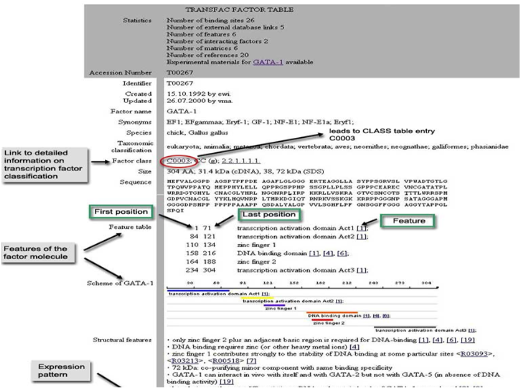 Figure 2. Transcription factor feature. Transcription factor의 서열 정보, 종 정보, 조직 정보, 도메인 정보, binding site 정보, interaction protein 정보, regulation정보를 총체적으로 서비스하고 있다.
Figure 2. Transcription factor feature. Transcription factor의 서열 정보, 종 정보, 조직 정보, 도메인 정보, binding site 정보, interaction protein 정보, regulation정보를 총체적으로 서비스하고 있다.
GO category정보 및 pathway정보도 가능한 모두 서비스가 되고 있어 세포내 생물학적 기능을 종합적으로 분석하고자 할 때 기초자료로 많은 정보를 주고 있다(figure3).
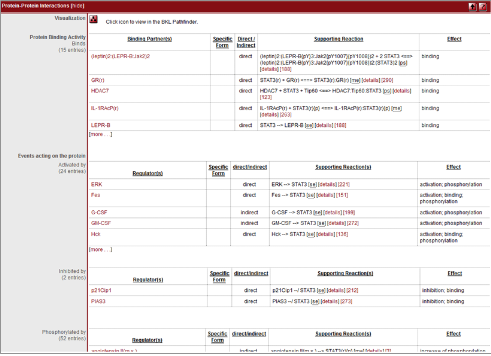 Figure 3. Transcription factor의 function 정보. Factor간의 interaction정보, pathway 정보, inhibitor 및 activator와 같은 regulation 정보 등을 문헌자료를 통해 데이터베이스화하고 서비스한다.
Figure 3. Transcription factor의 function 정보. Factor간의 interaction정보, pathway 정보, inhibitor 및 activator와 같은 regulation 정보 등을 문헌자료를 통해 데이터베이스화하고 서비스한다.
GO category정보 및 pathway정보도 가능한 모두 서비스가 되고 있어 세포내 생물학적 기능을 종합적으로 분석하고자 할 때 기초자료로 많은 정보를 주고 있다(figure3).
미지의 서열에 binding 가능한 transcription factor search.
특정한 발현 패턴을 보이는 유전자의 발현 조절 메커니즘을 분석 하고자 할 때 기본적으로 유전자의 upstream 영역에서 작용하는 transcription factor(TF)를 알아보게 된다. TRNASFAC®은 기본적인 transcription factor 및 binding site에 대한 정보를 제공함과 동시에 미지 서열에 binding 가능한 transcription factor를 예측할 수 있는 MatchTM, PatchTM, 그리고 Catch® 프로그램도 제공하고 있다(Figure 4).
MatchTM는 TF의 binding site를 matrix로 구성하여 찾는 방법이며, PatchTM는 서열의 pattern match 방법을 이용하여 찾는 방법이다. Catch®는 composite elements를 찾고자 할 때 사용하게 되는데 보통 이들 프로그램을 모두 사용하여 가능한 모든 TF를 찾고 실험에 이용한다. 또한 실험적으로 하나하나 규명할 수도 있으나 유전체 전체 유전자를 대상으로 분석하고자 할 때, 웹으로 운영되는 다음 프로그램에 서열을 하나씩 분석하기는 매우 어려우므로 local 서버나 PC에 설치하여 batch로 서열을 분석할 수도 있다. 이후 얻어진 유전자의 upstream 영역에서 작용하는 TF의 profile정보는 통계적 기법을 통해 유의한 TF를 선별하기도 하고, 데이터베이스화하기도 한다.
또한 얼마 전 덴마크의 CLCBio사와의 협력을 통해 CLCMainWorkbench 혹은 CLCGenomicsWorkbench의 plug-in 기능을 통해 TF정보를 visualization 할 수도 있게 되었다. 따라서 NGS에 의한 RNA-seq 정보 및 유전자 발현정보와 함께 전사조절 , 메커니즘까지 확대하여 함께 분석할 수 있는 최적의 데이터를 제공하고 있는 것이다.
Posted by 人Co
- Tag
- binding sites, Catch, CHIP-Seq, CLC Genomics Workbench, CLC Main Workbench, Expression, GO category, insilicogen, mamalian, Match TM, microarray, miRNA, NGS, Patch TM, pathway, Plant, Promoter, Reference, regulation, RNA polymerase, Species, TF, Transcription factor, TRANSFAC, TRANSFAC Professional, upstream, 도메인, 인실리코젠
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/71