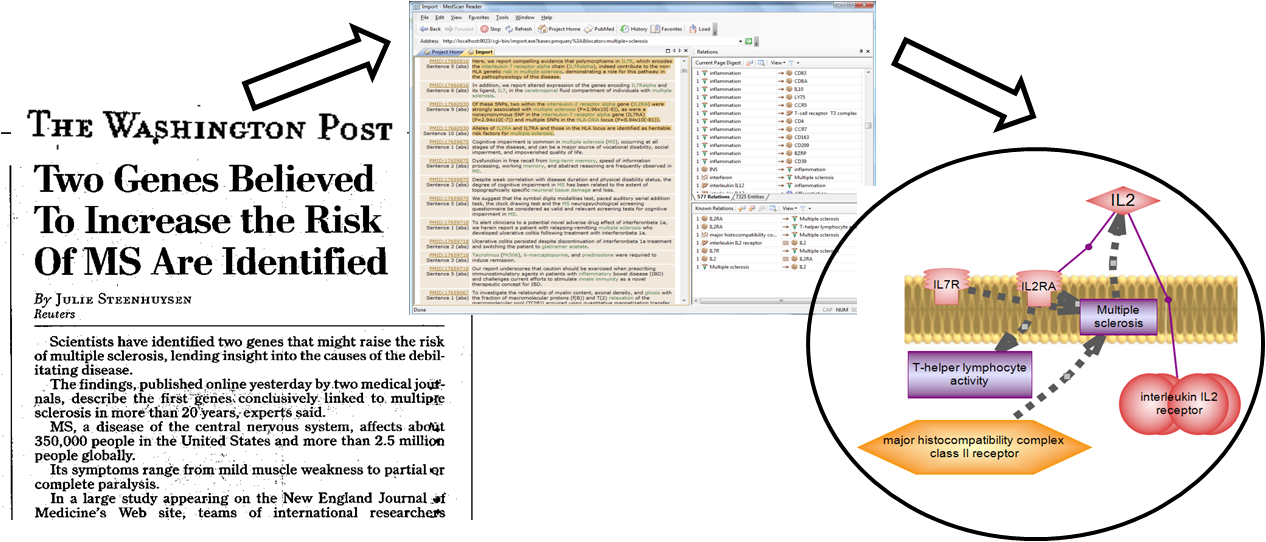
문화가 아름다운 회사 Insilicogen CultureDay!
- Posted at 2010/08/27 16:03
- Filed under 회사소식

문화가 아름다운 회사 Insilicogen의 CultureDay!
Insilicogen의 직원님들과 함께 동료애와 친목을 돈독히 할 수 있는 CultureDay를 소개합니다.
1월, 3월, 5월, 7월, 9월, 11월 넷째주 수요일 저녁에 핫이슈 영화를 다 같이 관람하는 날입니다.
물론 맛있는 저녁식사와 함께~
일주일 전, 이슈영화를 꼼꼼히 골라서 여러분들의 의견을 모아 영화를 선정합니다.
CultureDay영화가 공지되면 참석여부를 결정하고 참석희망자를 체크하여
서둘러 좋은 자리로 예매 완료!
넷째주 수요일 저녁이 되면 모두 모여서 맛있는 저녁식사를 하고 흥미진진한 영화도 관람합니다. CultureDay! 너무 매력적이지 않나요?
그럼 지금부터 지난 7월 CultureDay이야기를 담아보겠습니다.
7월에는 기대되는 개봉 영화가 많이 있었습니다.
파괴된사나이, 이끼, 마음이, 인셉션 등등..
영화를 선정하기가 고민스러웠지만 지금까지도 HOT한 영화가 있습니다.
참석희망자분들의 만장일치로 선정된 인셉션!

생각을 훔쳐라 VS. 생각을 지켜라
드림머신이라는 기계로 타인의 꿈과 접속해 생각을 빼낼 수 있는 미래사회. ‘돔 코브’(레오나르도 디카프리오)는 생각을 지키는 특수보안요원이면서 또한 최고의 실력으로 생각을 훔치는 도둑이다. 우연한 사고로 국제적인 수배자가 된 그는 기업간의 전쟁 덕에 모든 것을 되찾을 수 있는 기회를 얻게 된다. 하지만 임무는 머릿속의 정보를 훔쳐내는 것이 아니라, 반대로 머릿속에 정보를 입력시켜야 하는 것! 그는 ‘인셉션’이라 불리는 이 작전을 성공시키기 위해 최강의 팀을 조직한다. 불가능에 가까운 게임, 하지만 반드시 이겨야만 한다!
말 그대로 신비한 꿈의 세계 내용입니다. 생각을 훔칠 수 있는 거대한 전쟁.
인셉션은 드림머신이라는 기계로 타인의 꿈과 접속해 생각을 빼낼 수 있는 미래사회 이야기로 내용은 조금 복잡하고 어렵지만 중간 중간 이해가지 않는 내용이 있어도 집중해서 보면 아주 흥미진진합니다.

의뢰인의 요청에 구성된 팀은 본격적인 작전에 돌입. 의뢰인이 요청한 어느 대기업의 후계자의 기억에 침투하는 작전을 시작합니다. 꿈속에서는 ‘킥’을 통해 현실 세계로 돌아올 수 있는데 여기서 킥이란 높은 곳에서 아래로 떨어지거나 꿈속에서의 죽음 또는 강한 충격을 의미합니다. 이번 꿈은 꿈속의, 꿈속의, 꿈속의, 꿈속의, 꿈속에서의 작전. 영화 후반부에서는 약물로 인해 킥이 제대로 작용하지 않아 위험한 일을 초래하게 됩니다.


작전의 성공여부가 확연히 드러나지 않은 상태에서 처음 작전을 시작하게 된 기내로 돌아와 각각의 멤버는 알 수 없는 표정을 지었고, 현상수배범이었던 침입자는 유유히 미국에 입국하여 그토록 간절히 보고 싶어 하는 아이들을 만납니다. 본인 자신도 아직 꿈인지 현실인지 알 수 없는 마지막 결말부에서 그 답을 알려주는 열쇠 ‘토템’을 돌리고 아이들과 만날 수 있게 됩니다.

토템은 멈추면 현실, 멈추지 않으면 꿈이라는 것을 증명하는데 토템이 위태롭게 흔들리며 끝내 멈추지는 않은 채 영화는 막을 내립니다.

토템은 멈출까요? 계속 돌까요?
가장 성공적인 영화는 엔딩크레딧이 올라간 뒤에도 관객들로 하여금 끊임없이 얘기하게 만드는 영화라고 하는데 그런 점에서 크리스토퍼 놀란 감독은 완벽하게 성공한 것 같습니다.
꿈일까, 현실일까.
열린 결말의 기대 이상, 상상 이상의 작품.
사장님의 적극적인 지원으로 정말 즐겁고 행복했던 CultureDay!
직원을 위한 배려에 감사함을 느끼면서 우리의 인연을 더욱 소중히 하는 계기가 되었습니다.
다음 CultureDay를 기대해보며... ^^
추가로 디자이너들 입장에서 그린 인셉션 포스터를 소개합니다~ 즐거운 눈으로 감상해주세요!

출처:http://www.mymodernmet.com/profiles/blogs/10-fantastic-inception
by Descign team designer 찌:야♥
Posted by 人Co
- Tag
- 2010, CultureDay, Dinner, insilicogen, Movie, 인셉션, 인실리코젠
- Response
- No Trackback , 3 Comments
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/80