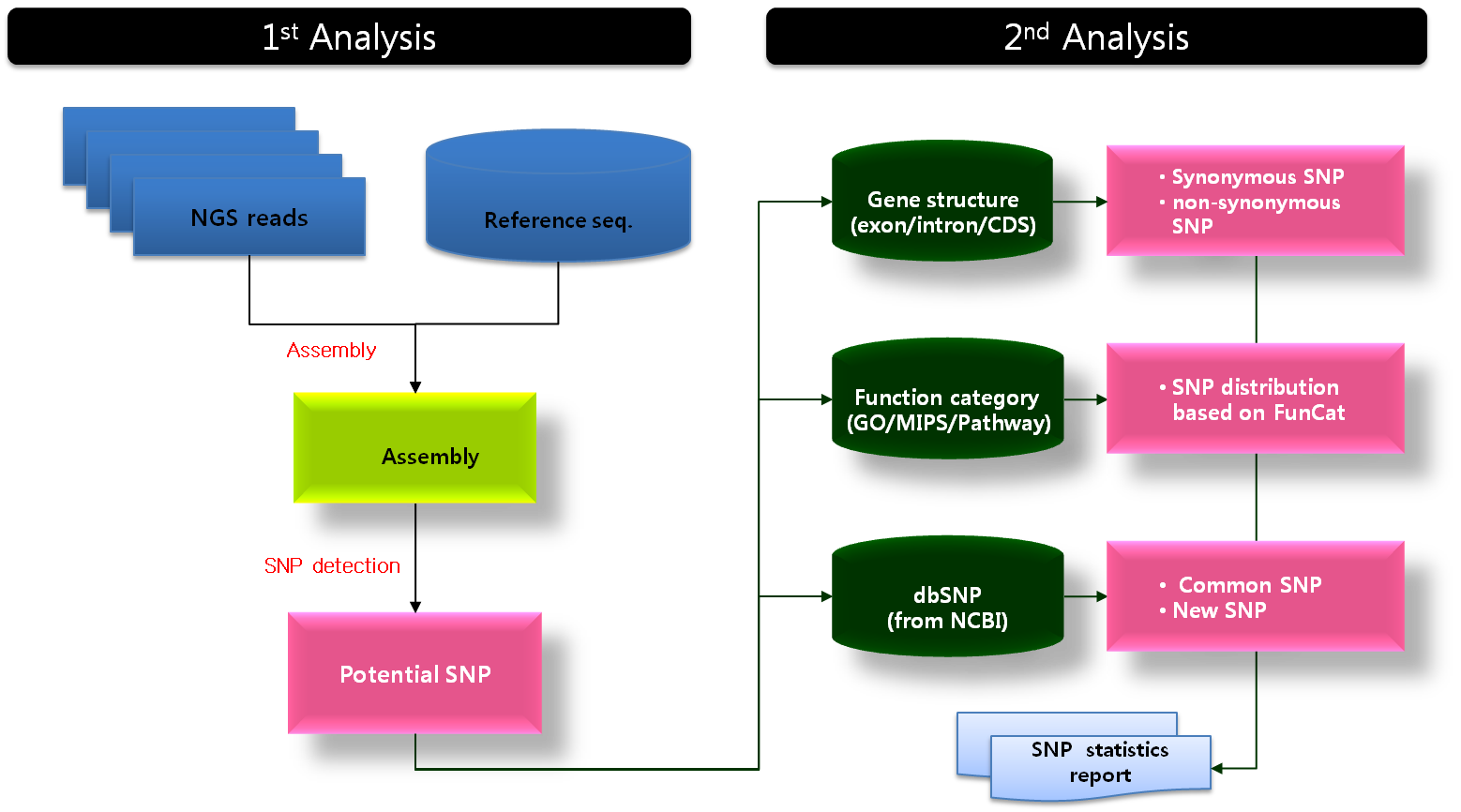
아니 대체 시퀀싱으로 어떻게 발현량을 알 수 있다는거야?
- Posted at 2012/04/30 20:10
- Filed under 제품소식
RNA-Seq
RNA-Seq은 NGS 기술로 transcriptome을 분석 할 수 있는 방법으로써, 말 그대로 특정 샘플에서 발현되는 RNA 서열을 시퀀싱하여, 어떤 exon들로 조합된 transcript가 발현이 되었는지, transcriptome에 대한 다양한 정보를 한 번에 알아낼 수 있는 방법입니다.
RNA-Seq 데이터 다운받기
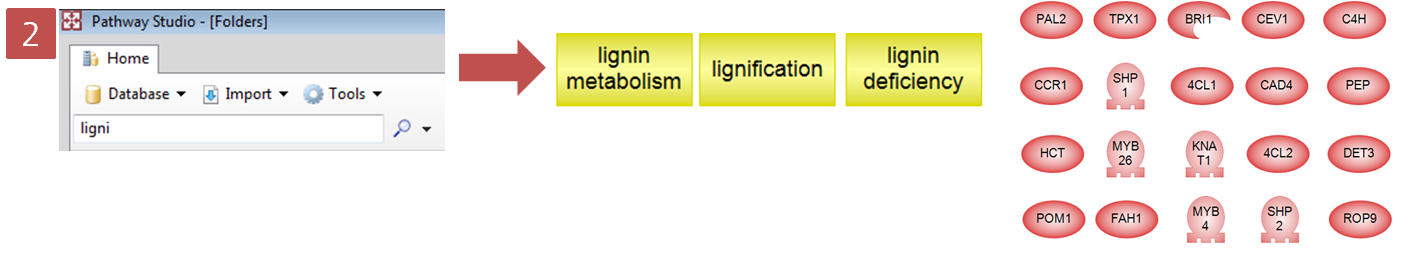
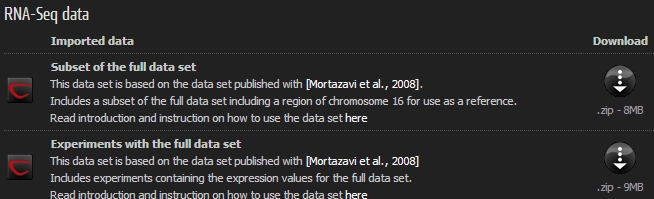
웹 브라우저에서 아래 url로 이동해 보시면 CLC bio에서 제공하는 예제 RNA-Seq 데이터를 받으실 수 있습니다. 이 데이터는 RNA-Seq 분석에 관한 초기 논문 중의 하나인 Mortazavi의 논문에서 얻은 데이터로 mouse의 brain과 liver에서 발현되는 mRNA를 시퀀싱 하여 분석한 데이터입니다. 이 데이터는 이미 CLC Genomics Workbench에 import가 된 상태의 데이터로 .zip 파일을 그대로 ‘Navigation Area'에 드래그 하면 자동으로 import가 됩니다.
http://www.clcbio.com/index.php?id=1290
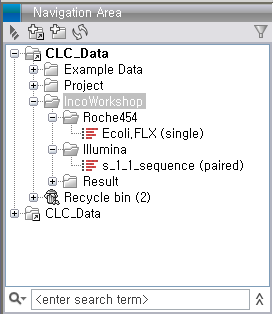
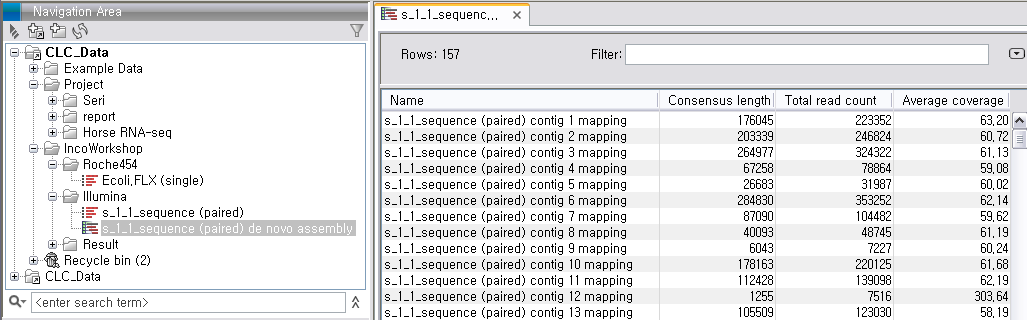
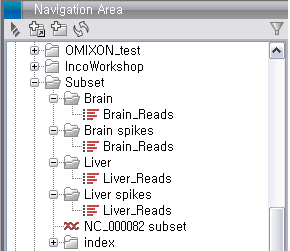
 Subset.zip 이라는 파일의 import가 완료되면 다음 그림과 같은 데이터가 나타나게 됩니다. Brain과 liver에서
각각 두 개씩 얻은 read 데이터와 mouse 16번 염색체의 reference 서열 파일을 확인 할 수 있습니다.
Subset.zip 이라는 파일의 import가 완료되면 다음 그림과 같은 데이터가 나타나게 됩니다. Brain과 liver에서
각각 두 개씩 얻은 read 데이터와 mouse 16번 염색체의 reference 서열 파일을 확인 할 수 있습니다.
RNA-Seq 분석 돌리기
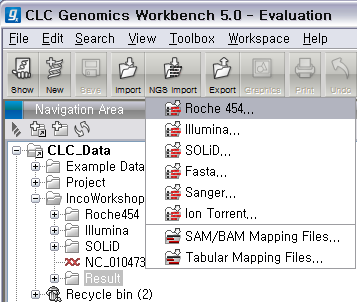
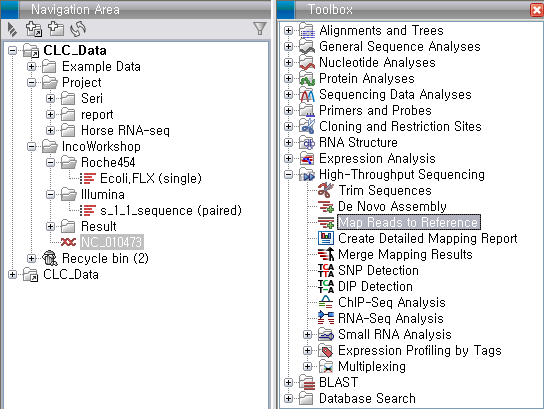
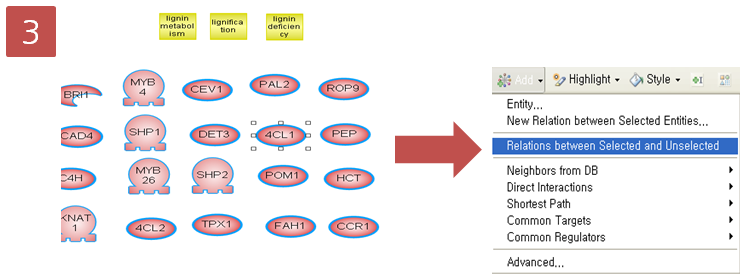
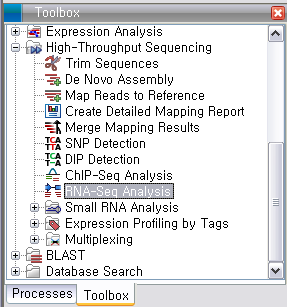
데이터 준비가 완료되면, ‘Toolbox’의 'RNA-Seq Analysis'를 실행시킵니다.
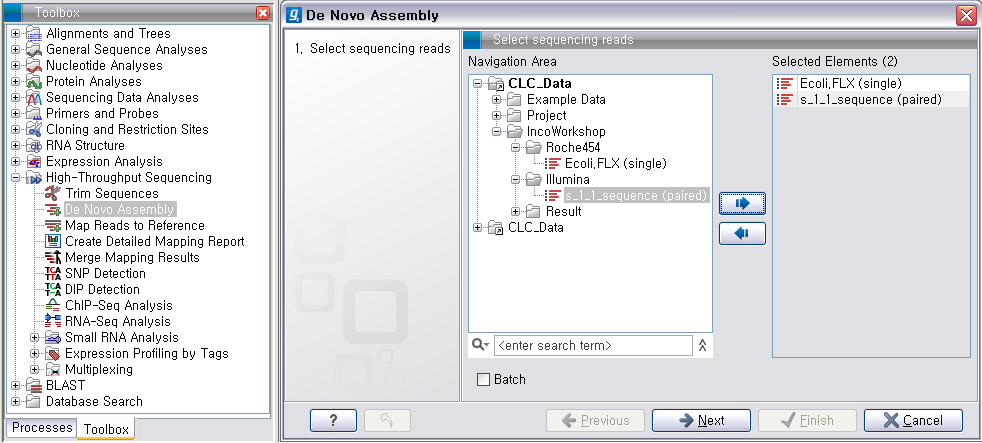
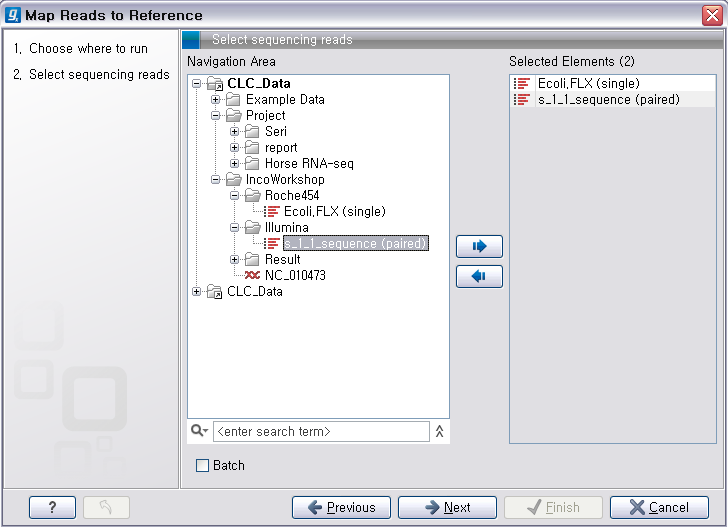
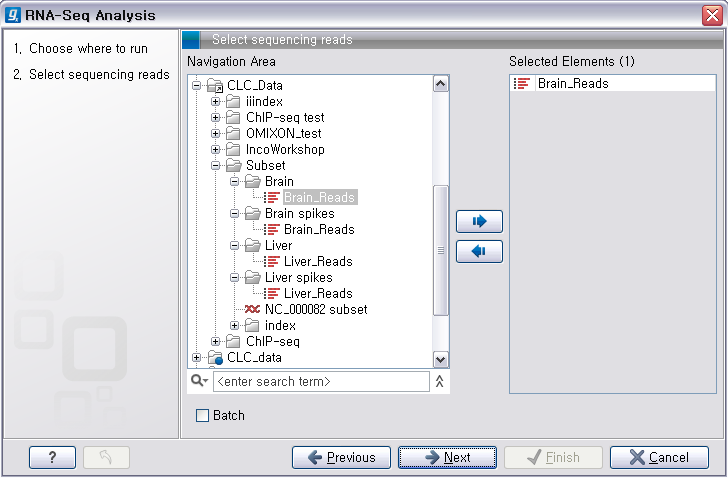
 첫 번째 단계에서 reference 서열에 mapping 시킬 read 데이터를 선택합니다. 이때 각 샘플에서 얻은 데이터끼리 따로 분석을 해야 하기 때문에, 다른 샘플의 read를 함께 선택하지 않도록 주의합니다.
첫 번째 단계에서 reference 서열에 mapping 시킬 read 데이터를 선택합니다. 이때 각 샘플에서 얻은 데이터끼리 따로 분석을 해야 하기 때문에, 다른 샘플의 read를 함께 선택하지 않도록 주의합니다.
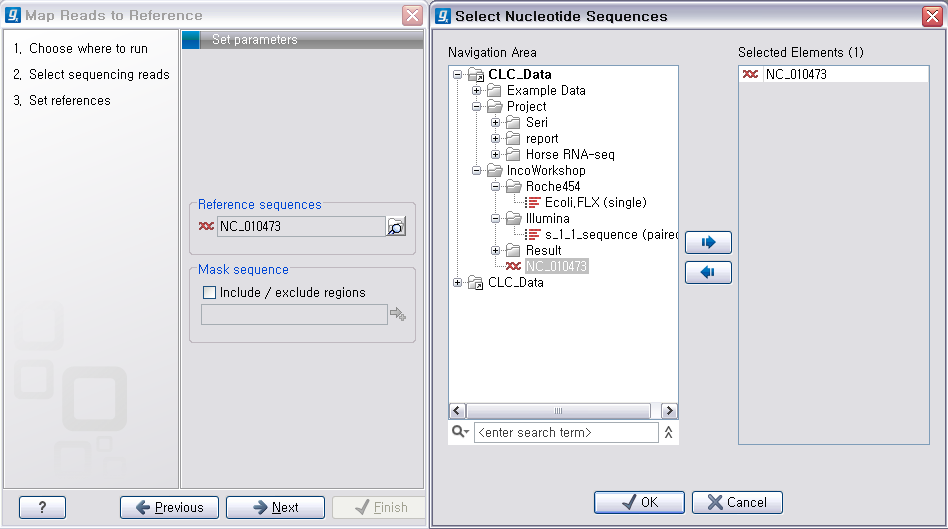
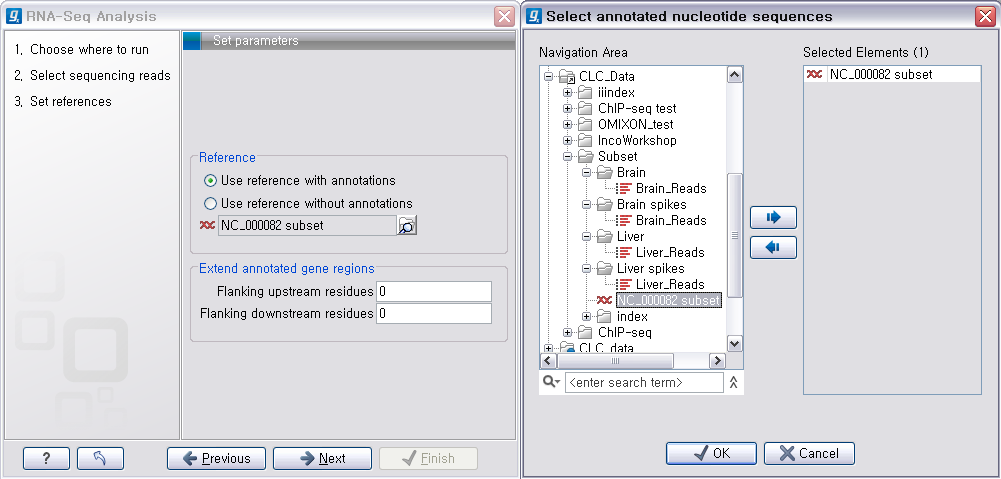
 다음 단계에서는 reference 서열에 대한 몇 가지 항목을 설정합니다. ‘Reference'는 read를 mapping 시킬
reference 서열을 지정하는 항목인데 선택한 reference 서열에 있는 annotation을 이용할 것인지 아닌지를
선택해야 합니다. 전자의 경우 reference 서열에서 'Gene'이라는 이름으로 annotation 영역을 추출한 다음 그
서열들에만 read들을 mapping 시키게 됩니다. 이 때 아래 쪽 ’Extend annotated gene regions'의
값을 조정하면 gene 영역의 upstream과 downstream으로 지정된 base 만큼 확장하여 추출하게 됩니다. 후자의
경우 전체 reference 서열에 read들을 mapping 시킨 후 전체 서열에 대한 발현량이 계산되게 됩니다.
다음 단계에서는 reference 서열에 대한 몇 가지 항목을 설정합니다. ‘Reference'는 read를 mapping 시킬
reference 서열을 지정하는 항목인데 선택한 reference 서열에 있는 annotation을 이용할 것인지 아닌지를
선택해야 합니다. 전자의 경우 reference 서열에서 'Gene'이라는 이름으로 annotation 영역을 추출한 다음 그
서열들에만 read들을 mapping 시키게 됩니다. 이 때 아래 쪽 ’Extend annotated gene regions'의
값을 조정하면 gene 영역의 upstream과 downstream으로 지정된 base 만큼 확장하여 추출하게 됩니다. 후자의
경우 전체 reference 서열에 read들을 mapping 시킨 후 전체 서열에 대한 발현량이 계산되게 됩니다.
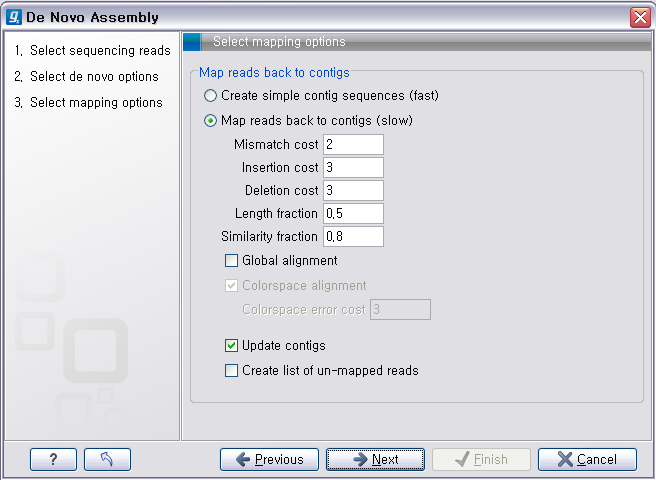
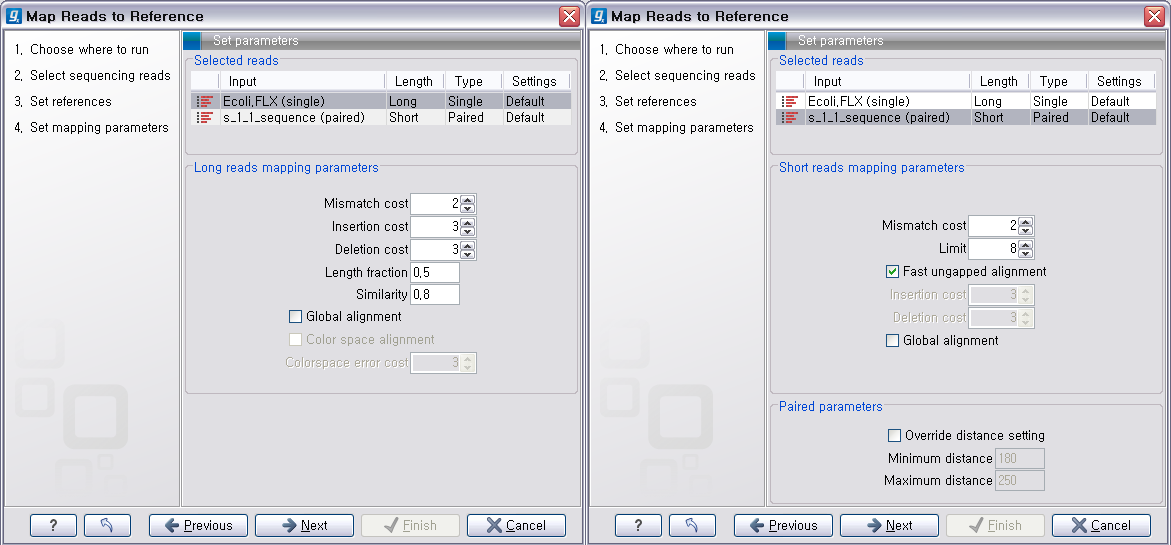
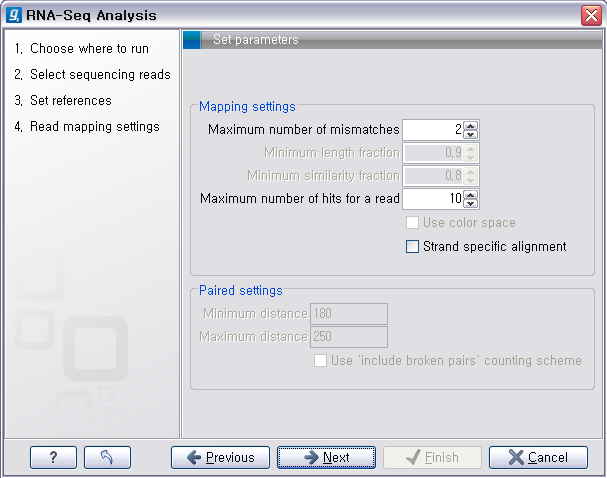
 다음 단계에서는 read를 mapping 하는데 요구되는 옵션들을 설정하게 됩니다. ‘Maximum number of
mismatches'는 read가 reference 서열에 mapping 될 때 허용되는 mismatch base의 수를 정해주는
옵션이고 ’Maximum number of hits for a read'는 non-specific하게 mapping 되는
read의 허용 가능한 정도를 정하는 옵션입니다. 예를 들어 이 옵션이 ‘10’으로 설정되어 있을 경우, reference 서열에
mapping 될 수 있는 부분이 11개 이상인 read는 mapping되지 않고 버려집니다. 반면에 mapping 될 수 있는
부분이 10개 이하인 경우에는 그 mapping 될 수 있는 부분들 중에서 무작위로 한 자리가 선택되어 mapping 되게
됩니다. 'Minimum length fraction'과
‘Minimum similarity fraction'은 mapping 시킬 read가 long read 일 경우 적용되는
옵션입니다. Long read는 길이가 길다 보니 reference 서열과 mapping 되는 부분을 base 단위로 정하지 않고
비율로 정하게 되는데, 'Minimum length fraction'이 ’0.9‘로 설정되면 100bp의 read는 최소한
90bp 이상 reference 서열과 match되어야 mapping 됩니다. 그리고 ‘Minimum similarity
fraction'이 ’0.8‘로 설정되면 mapping 된 부분의 identity가 80%는 되어야 mapping이 됩니다.
다음 단계에서는 read를 mapping 하는데 요구되는 옵션들을 설정하게 됩니다. ‘Maximum number of
mismatches'는 read가 reference 서열에 mapping 될 때 허용되는 mismatch base의 수를 정해주는
옵션이고 ’Maximum number of hits for a read'는 non-specific하게 mapping 되는
read의 허용 가능한 정도를 정하는 옵션입니다. 예를 들어 이 옵션이 ‘10’으로 설정되어 있을 경우, reference 서열에
mapping 될 수 있는 부분이 11개 이상인 read는 mapping되지 않고 버려집니다. 반면에 mapping 될 수 있는
부분이 10개 이하인 경우에는 그 mapping 될 수 있는 부분들 중에서 무작위로 한 자리가 선택되어 mapping 되게
됩니다. 'Minimum length fraction'과
‘Minimum similarity fraction'은 mapping 시킬 read가 long read 일 경우 적용되는
옵션입니다. Long read는 길이가 길다 보니 reference 서열과 mapping 되는 부분을 base 단위로 정하지 않고
비율로 정하게 되는데, 'Minimum length fraction'이 ’0.9‘로 설정되면 100bp의 read는 최소한
90bp 이상 reference 서열과 match되어야 mapping 됩니다. 그리고 ‘Minimum similarity
fraction'이 ’0.8‘로 설정되면 mapping 된 부분의 identity가 80%는 되어야 mapping이 됩니다.
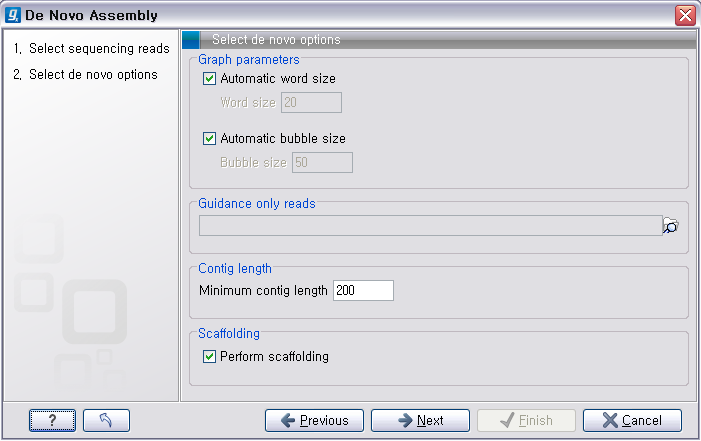
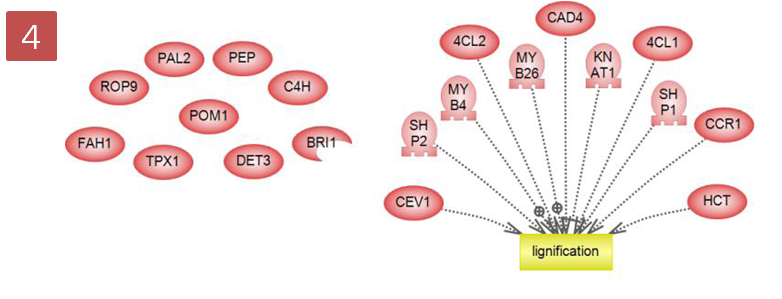
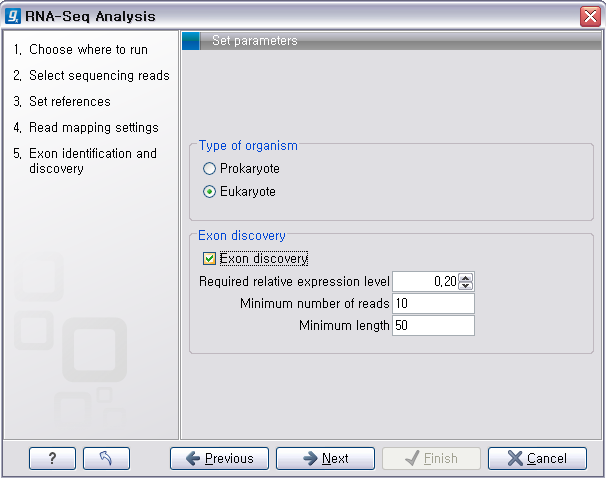
 다음 옵션은 새로운 exon 영역을 찾아내는데 필요한 옵션들입니다. 먼저 'Type of organism'에서 분석 대상이
원핵생물(Prokaryote)인지 진핵생물(Eukaryote)인지 선택합니다. 원핵생물의 경우 exon과 intron의 개념이
없기 때문에 'Exon discovery'가 수행되지 않습니다.
다음 옵션은 새로운 exon 영역을 찾아내는데 필요한 옵션들입니다. 먼저 'Type of organism'에서 분석 대상이
원핵생물(Prokaryote)인지 진핵생물(Eukaryote)인지 선택합니다. 원핵생물의 경우 exon과 intron의 개념이
없기 때문에 'Exon discovery'가 수행되지 않습니다.
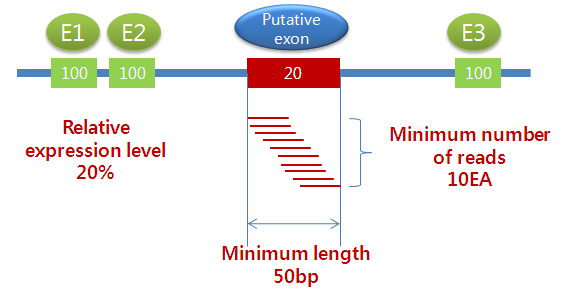
 진핵생물을 선택하고 'Exon discovery'를 수행하도록 체크하게 되면, 세 가지 옵션 값을 설정 할 수 있습니다.
'Required relative expression level'은 다른 exon들의 발현량에 비해서 새롭게 찾아진 exon에
요구되는 상대적인 발현량을 의미합니다. 그리고 ‘Minimum number of reads'는 새롭게 찾아진 exon 영역에
요구되는 최소한의 mapping read의 수를 의미하고, 'Minimum length'는 그 exon 영역의 최소 길이를
의미합니다. 예를 들어 이 옵션들이 기본 값으로 설정된 경우, intron 영역의 어떤 부분에 10개 이상의 read가
mapping 되고, 이 read 들로 조합된 consensus 영역이 50bp 이상이면서, 이 부분에 대하여 계산된 발현량이
다른 exon 들의 발현량에 대하여 상대적으로 20% 이상이면 이 영역을 기존에 알려지지 않은 새로운 exon 이라고 인식하도록
되어 있습니다.
진핵생물을 선택하고 'Exon discovery'를 수행하도록 체크하게 되면, 세 가지 옵션 값을 설정 할 수 있습니다.
'Required relative expression level'은 다른 exon들의 발현량에 비해서 새롭게 찾아진 exon에
요구되는 상대적인 발현량을 의미합니다. 그리고 ‘Minimum number of reads'는 새롭게 찾아진 exon 영역에
요구되는 최소한의 mapping read의 수를 의미하고, 'Minimum length'는 그 exon 영역의 최소 길이를
의미합니다. 예를 들어 이 옵션들이 기본 값으로 설정된 경우, intron 영역의 어떤 부분에 10개 이상의 read가
mapping 되고, 이 read 들로 조합된 consensus 영역이 50bp 이상이면서, 이 부분에 대하여 계산된 발현량이
다른 exon 들의 발현량에 대하여 상대적으로 20% 이상이면 이 영역을 기존에 알려지지 않은 새로운 exon 이라고 인식하도록
되어 있습니다.
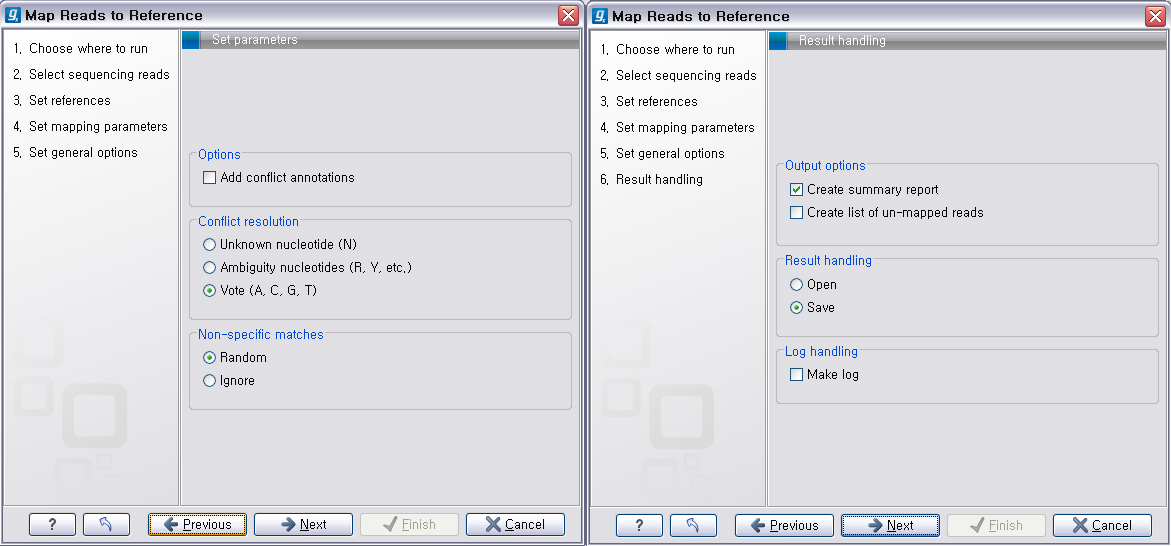
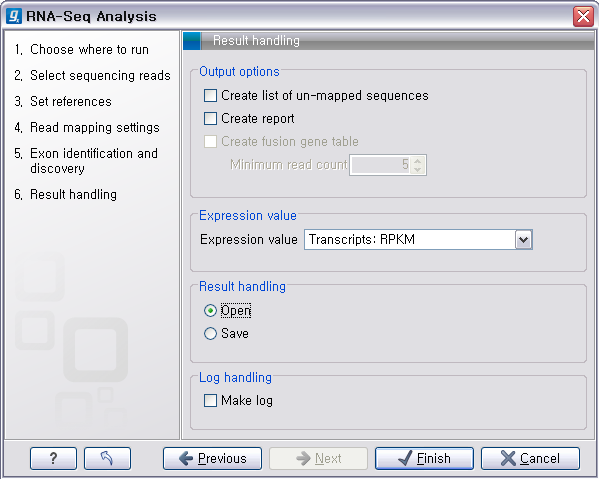
 다음 단계에서는 분석 결과를 작성하는데 필요한 몇 가지 옵션들을 설정하게 됩니다. Mapping 되지 않은 read들의 목록을
따로 생성시킬 것인지, RNA-seq 분석에 관한 report나 분석 log를 작성할 것인지에 관하여 설정할 수 있습니다.
'Expression value'는 각 유전자 혹은 transcript의 발현값을 어떻게 계산 할 것인지를 정하는 옵션입니다.
'Transcript:RPKM'을 선택하면 각 transcript의 발현값을 계산하여 보여지게 됩니다.
다음 단계에서는 분석 결과를 작성하는데 필요한 몇 가지 옵션들을 설정하게 됩니다. Mapping 되지 않은 read들의 목록을
따로 생성시킬 것인지, RNA-seq 분석에 관한 report나 분석 log를 작성할 것인지에 관하여 설정할 수 있습니다.
'Expression value'는 각 유전자 혹은 transcript의 발현값을 어떻게 계산 할 것인지를 정하는 옵션입니다.
'Transcript:RPKM'을 선택하면 각 transcript의 발현값을 계산하여 보여지게 됩니다.
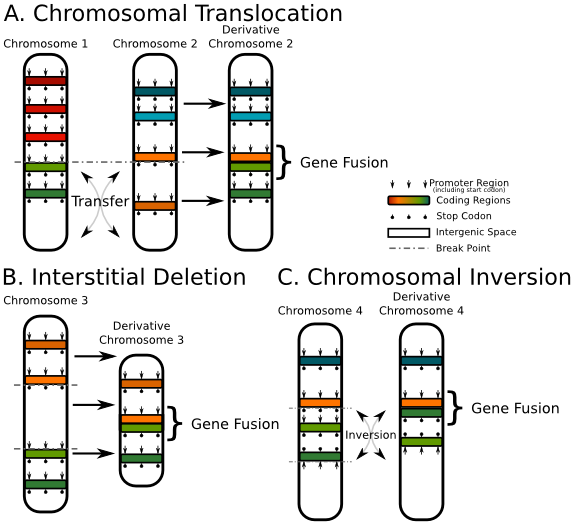
 그리고 paired-end read를 사용할 경우 'gene fusion' 분석도 할 수 있습니다. Gene fusion은
translocation, deletion, inversion과 같이 염색체 구조 변이에 의해서 두 개의 유전자가 합쳐진 경우를
말합니다.
그리고 paired-end read를 사용할 경우 'gene fusion' 분석도 할 수 있습니다. Gene fusion은
translocation, deletion, inversion과 같이 염색체 구조 변이에 의해서 두 개의 유전자가 합쳐진 경우를
말합니다.
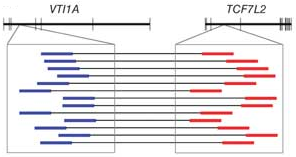
 예를 들어, gene fusion이 일어난 유전자에서 mRNA가 발현되고 시퀀싱 하여 paired-end read를 얻은 후
정상적인 reference 서열에 mapping을 시켜보면 forward 서열은 A라는 유전자에 mapping 되는데
reverse 서열은 B라는 유전자에 mapping 될 수 있습니다.
예를 들어, gene fusion이 일어난 유전자에서 mRNA가 발현되고 시퀀싱 하여 paired-end read를 얻은 후
정상적인 reference 서열에 mapping을 시켜보면 forward 서열은 A라는 유전자에 mapping 되는데
reverse 서열은 B라는 유전자에 mapping 될 수 있습니다.
 이런 paired-end read를 두 유전자 사이에 gene fusion이 일어났다고 볼 수 있는 증거로 제시할 수 있으며,
‘Minimum read count'로 이런 paired-end read가 최소한 몇 개가 있어야 gene fusion이
일어났다고 report를 할지 정해 줄 수 있습니다.
이런 paired-end read를 두 유전자 사이에 gene fusion이 일어났다고 볼 수 있는 증거로 제시할 수 있으며,
‘Minimum read count'로 이런 paired-end read가 최소한 몇 개가 있어야 gene fusion이
일어났다고 report를 할지 정해 줄 수 있습니다.
모든 옵션과 결과의 저장 위치 지정이 완료되면 ‘Finish' 버튼을 클릭하면 분석이 진행 됩니다.
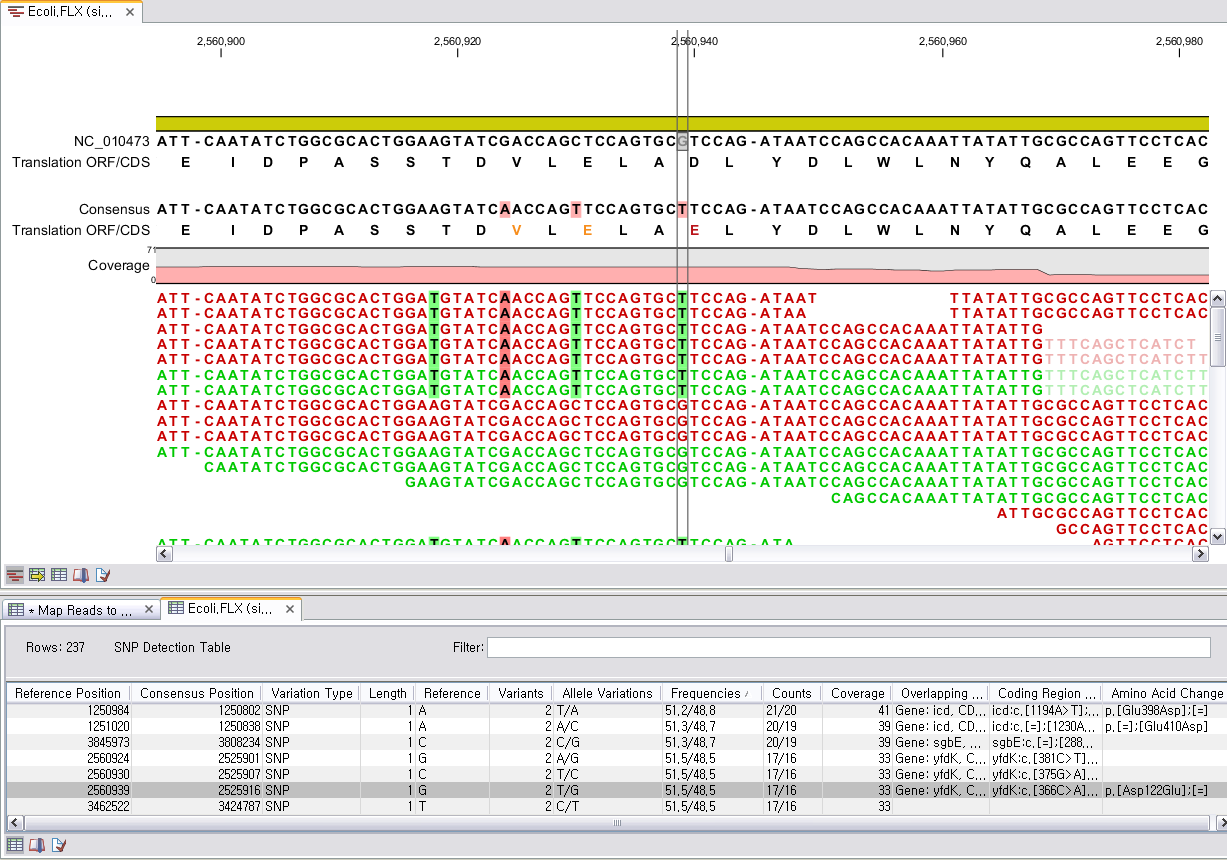
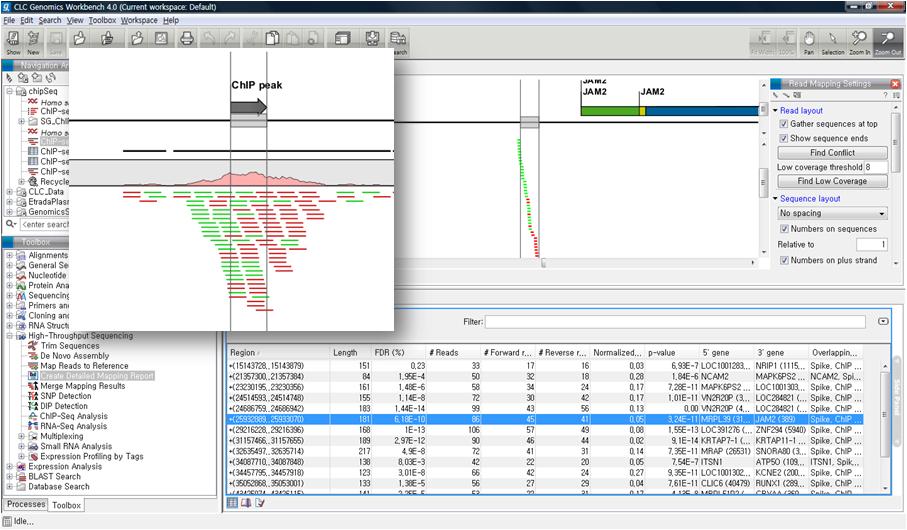
분석이 완료되면 다양한 정보가 들어있는 테이블이 나타납니다. 각 유전자 별로 발현값, annotate된 transcript의 수, 확인된 transcript의 수 exon 영역의 길이, exon 영역에 mapping된 read의 수, 등 많은 정보를 확인 할 수 있습니다.
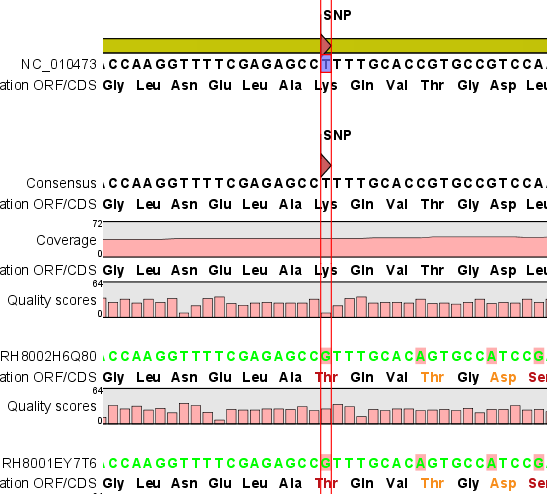
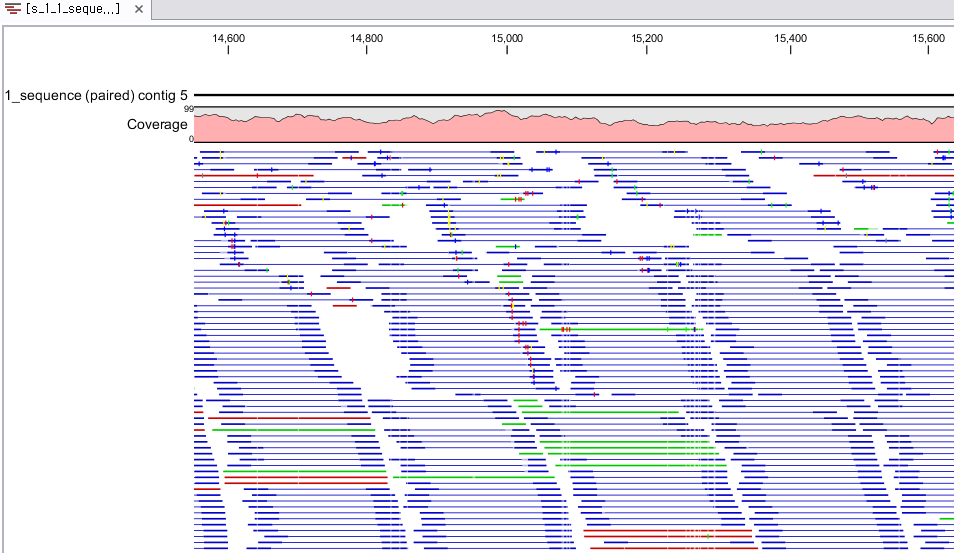
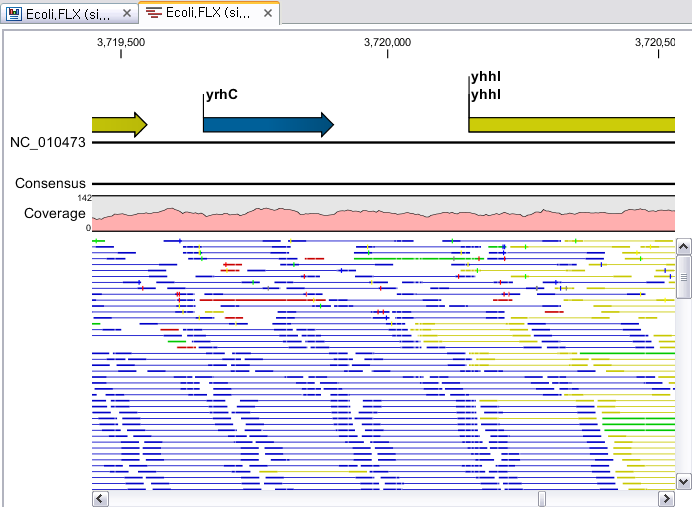
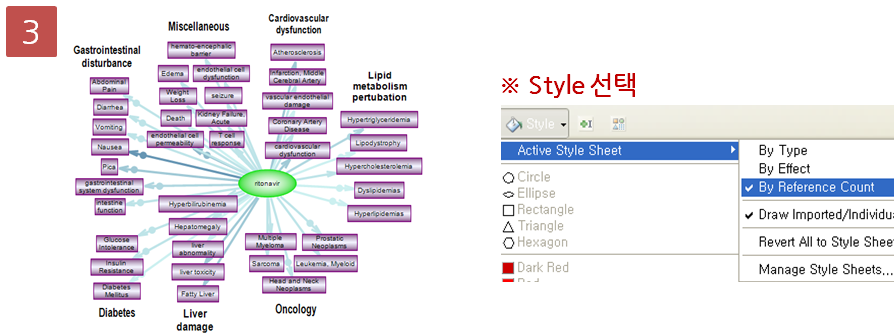
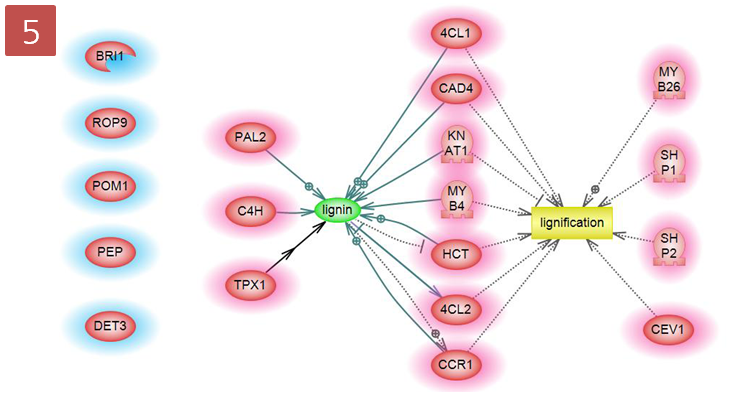
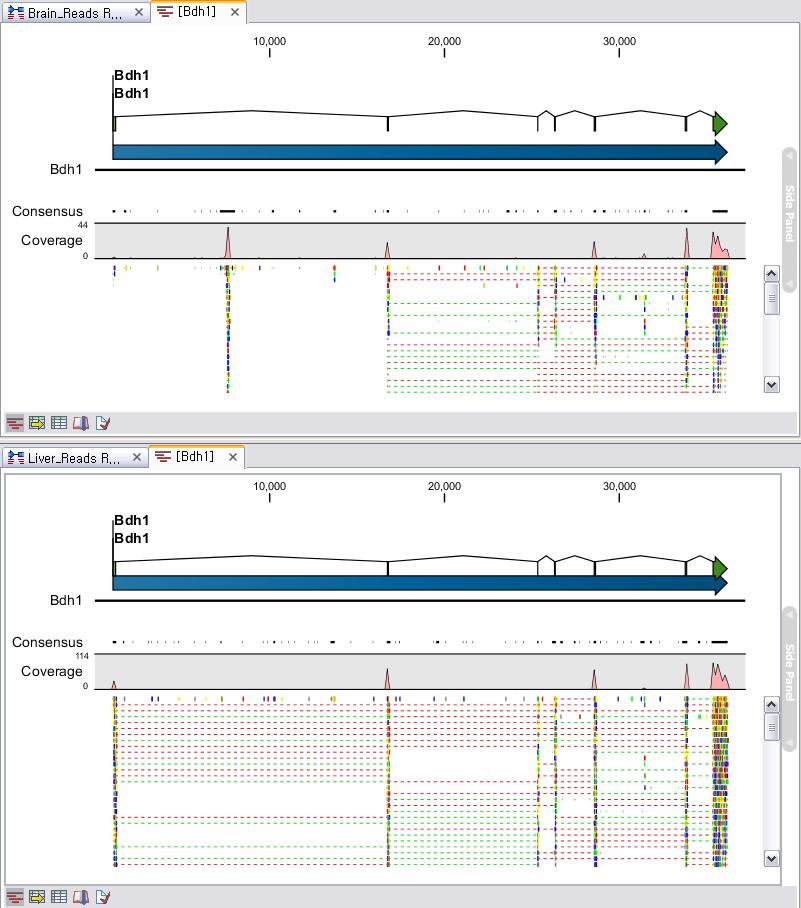
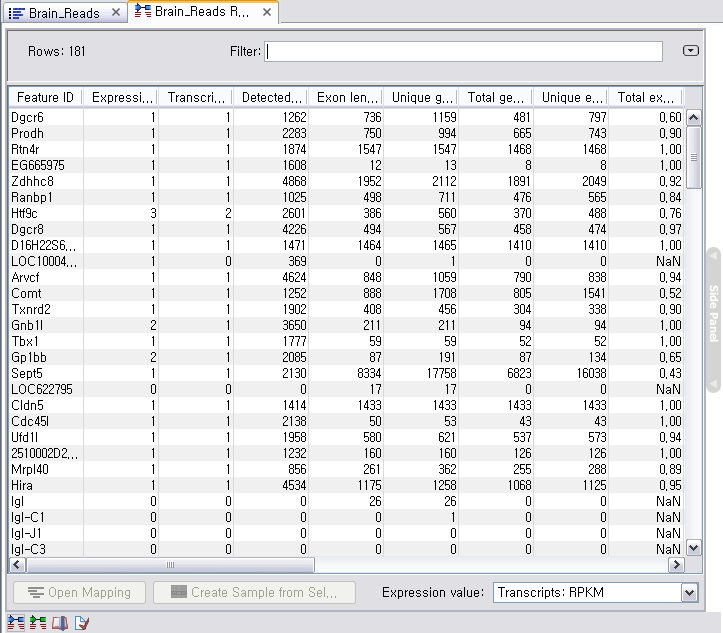 각 항목을 더블클릭해 보면 각 유전자별로 read 들이 어떻게 mapping 되었는지 확인할 수 있는 mapping view가
나타납니다. Mapping view에서 오른쪽 side panel의 몇 가지 설정을 바꾸면 다양한 형태로 화면을 수정할 수
있습니다. Mapping view를 열어 보면 read들이 어떤 exon에 mapping이 되었는지 볼 수 있고, 이를 통해 어떤
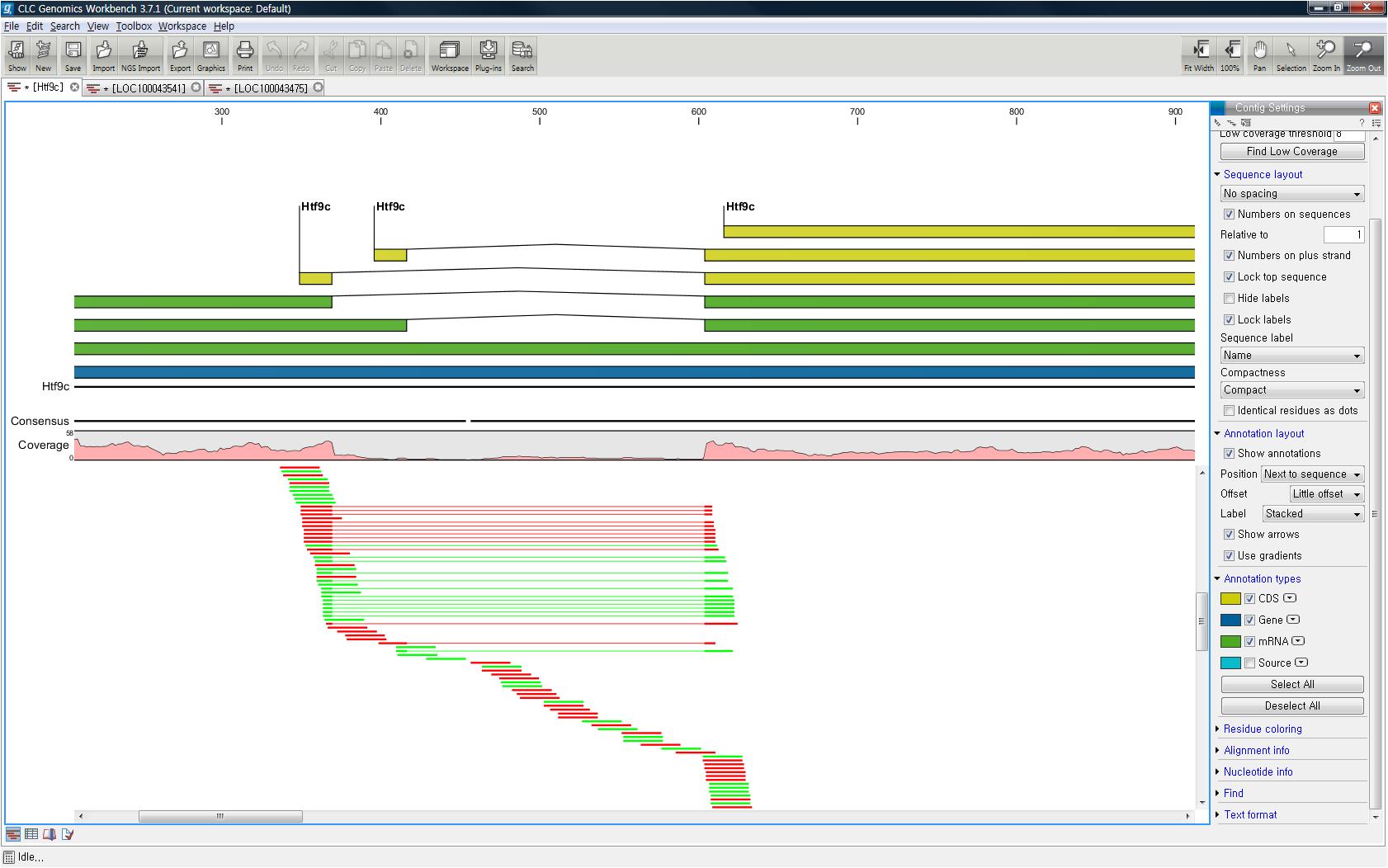
exon들이 조합된 transcript가 발현이 되었는지 알 수 있게 됩니다. 한 가지 예로 Brain sample의 Bdh1
이라는 유전자에서는 1개의 'Putative exon'이 발견 되었습니다. Liver sample의 결과에서 같은 유전자의
mapping view를 열고 비교해 보니 Brain sample에서 발견된 새로운 exon 부분이 mapping 되지 않은 것을
볼 수 있습니다. 이를 통해 Brain 조직에서는 Bdg1 유전자에 기존에 알려지지 않은 exon 영역이 존재하고 이 exon
영역이 함께 조합된 새로운 transcript가 발현된다고 추측해 볼 수 있습니다.
각 항목을 더블클릭해 보면 각 유전자별로 read 들이 어떻게 mapping 되었는지 확인할 수 있는 mapping view가
나타납니다. Mapping view에서 오른쪽 side panel의 몇 가지 설정을 바꾸면 다양한 형태로 화면을 수정할 수
있습니다. Mapping view를 열어 보면 read들이 어떤 exon에 mapping이 되었는지 볼 수 있고, 이를 통해 어떤
exon들이 조합된 transcript가 발현이 되었는지 알 수 있게 됩니다. 한 가지 예로 Brain sample의 Bdh1
이라는 유전자에서는 1개의 'Putative exon'이 발견 되었습니다. Liver sample의 결과에서 같은 유전자의
mapping view를 열고 비교해 보니 Brain sample에서 발견된 새로운 exon 부분이 mapping 되지 않은 것을
볼 수 있습니다. 이를 통해 Brain 조직에서는 Bdg1 유전자에 기존에 알려지지 않은 exon 영역이 존재하고 이 exon
영역이 함께 조합된 새로운 transcript가 발현된다고 추측해 볼 수 있습니다.
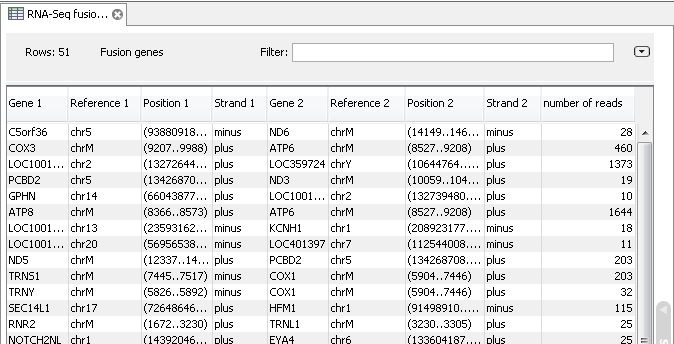
 Paired-end read를 이용하고 gene fusion event를 확인 하도록 옵션을 설정했다면, 다음과 같은 결과
테이블도 볼 수 있습니다. 이 테이블에서 gene fusion이 일어난 유전자와 그 유전자의 위치, 그리고 몇 개의
paired-end read가 mapping 되었는지 확인 할 수 있습니다.
Paired-end read를 이용하고 gene fusion event를 확인 하도록 옵션을 설정했다면, 다음과 같은 결과
테이블도 볼 수 있습니다. 이 테이블에서 gene fusion이 일어난 유전자와 그 유전자의 위치, 그리고 몇 개의
paired-end read가 mapping 되었는지 확인 할 수 있습니다.
Reference
- http://en.wikipedia.org/wiki/Fusion_gene
- Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion, Nature Genetics Volume:43, Pages:964–968 Year published:(2011)
* 아래 이메일 주소로 연락 주시면 CLC Genomics Workbench의 모든 기능을 사용할 수 있는 데모 라이센스를 제공해 드리오니 많은 이용 바랍니다.
- codes@insilicogen.com
RNA-Seq은 NGS 기술로 transcriptome을 분석 할 수 있는 방법으로써, 말 그대로 특정 샘플에서 발현되는 RNA 서열을 시퀀싱하여, 어떤 exon들로 조합된 transcript가 발현이 되었는지, transcriptome에 대한 다양한 정보를 한 번에 알아낼 수 있는 방법입니다.
RNA-Seq 데이터 다운받기
웹 브라우저에서 아래 url로 이동해 보시면 CLC bio에서 제공하는 예제 RNA-Seq 데이터를 받으실 수 있습니다. 이 데이터는 RNA-Seq 분석에 관한 초기 논문 중의 하나인 Mortazavi의 논문에서 얻은 데이터로 mouse의 brain과 liver에서 발현되는 mRNA를 시퀀싱 하여 분석한 데이터입니다. 이 데이터는 이미 CLC Genomics Workbench에 import가 된 상태의 데이터로 .zip 파일을 그대로 ‘Navigation Area'에 드래그 하면 자동으로 import가 됩니다.
http://www.clcbio.com/index.php?id=1290
RNA-Seq 분석 돌리기
데이터 준비가 완료되면, ‘Toolbox’의 'RNA-Seq Analysis'를 실행시킵니다.
모든 옵션과 결과의 저장 위치 지정이 완료되면 ‘Finish' 버튼을 클릭하면 분석이 진행 됩니다.
분석이 완료되면 다양한 정보가 들어있는 테이블이 나타납니다. 각 유전자 별로 발현값, annotate된 transcript의 수, 확인된 transcript의 수 exon 영역의 길이, exon 영역에 mapping된 read의 수, 등 많은 정보를 확인 할 수 있습니다.
Reference
- http://en.wikipedia.org/wiki/Fusion_gene
- Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion, Nature Genetics Volume:43, Pages:964–968 Year published:(2011)
* 아래 이메일 주소로 연락 주시면 CLC Genomics Workbench의 모든 기능을 사용할 수 있는 데모 라이센스를 제공해 드리오니 많은 이용 바랍니다.
- codes@insilicogen.com
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/109