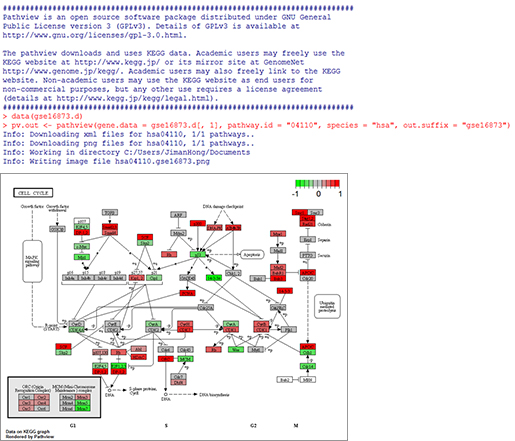
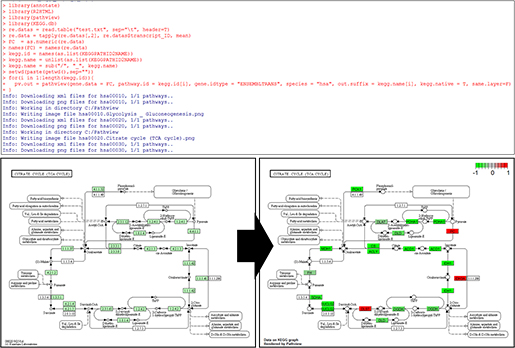
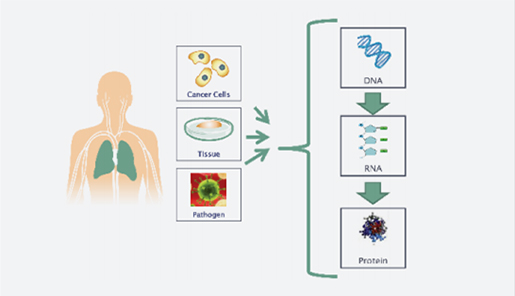
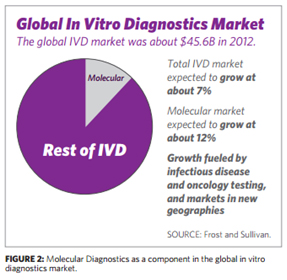
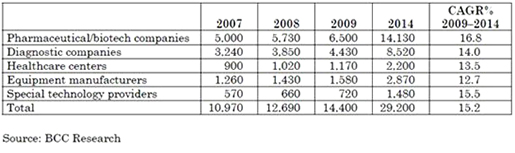
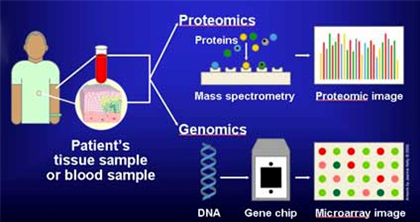
(주)인실리코젠 세계 최대규모 유방암 전장유전체 해독 연구 참여
- Posted at 2016/05/03 16:50
- Filed under 생물정보

<논문>
- Landscape of somatic mutations in 560 breast cancer whole-genome sequences
Nature (2016) doi:10.1038/nature17676
<관련기사>
- Breast cancer: Scientists hail 'very significant' genetic find BBC News
- ‘유방암 만드는 유전자’ 93개 밝혀졌다 경향신문
- 국내 연구진, 유방암 '전체 유전자 염기서열' 해독…네이처誌 발표 포커스뉴스
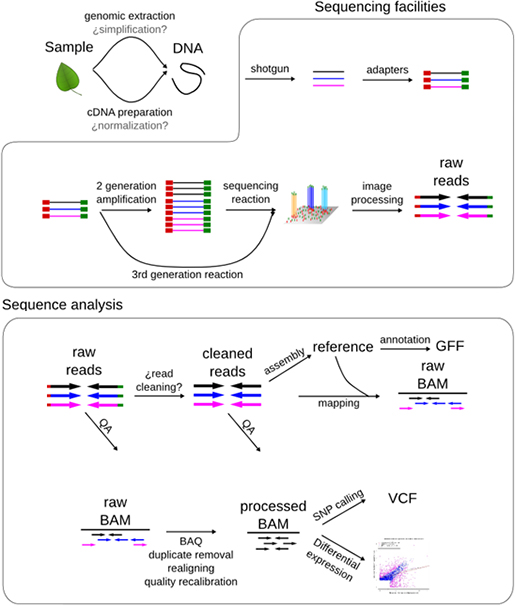
세계 최대규모의 유방암 전장유전체 해독 연구 결과가 네이처(Nature)를 통해 공개되었습니다. 이 연구는 한양대학교 의과대학 공구 교수팀과 영국 생어연구소(Sanger Institute) 암 유전체팀이 공동으로 주관하고, 12개국 48개 기관이 참여했습니다. 생물정보 전문기업 (주)인실리코젠의 김형용 수석개발자도 본 연구에 공동으로 참여했으며, 오랜 분석 기간과 리뷰 기간을 거쳐 오늘(5월 3일) 새벽 1시에 연구결과가 네이처 온라인판에 게재되었습니다.
이 연구는 최대규모의 암 전장유전체 분석(WGS, Whole genome sequencing)으로써, 국내외 유방암 환자 560명을 대상으로 암조직과 정상조직을 모두 전장유전체 해독하고, 정상조직과의 차이를 통해 암을 유발한 유전변이를 밝혔으며, 각 유전변이의 패턴을 규명하였습니다. 암은 발암물질, 자외선, 흡연 등 각종 원인으로 인한 유전변이의 누적으로 발생하는 질병이므로, 암을 일으키는 주요 유전변이의 완전한 해석은 발암 기전을 이해하고, 효과적인 치료방법을 제안하기 위해 매우 중요합니다. 또한, 암을 일으키는 유전변이는 환자 개인마다 매우 다양하게 나타나기 때문에 다수의 사례를 통해서만 종합적으로 이해할 수 있습니다. 이번 560 사례의 전장유전체 해독을 통해 유방암 유전변이와 발암 기전에 대해 좀 더 이해하고, 향후 암 정복을 위한 자세한 백과사전 역할을 수행할 수 있다는 점에서 그 의미가 크다고 할 수 있습니다.
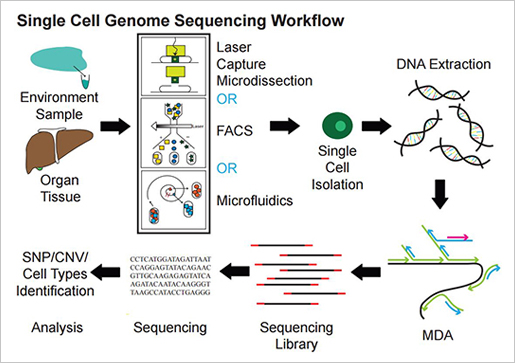
(주)인실리코젠의 김형용 수석개발자를 주축으로 SD그룹의 연구진들은 해독한 유전체의 데이터 기본 분석에 함께 참여했으며, 영국 생어연구소에 방문하여, 암 유전체팀의 전체 분석과정을 함께 리뷰할 수 있었습니다. CaVEMan, Pindel을 이용한 SNV, Indel 탐지, ASCAT을 이용한 copy number 분석, BRASS를 이용한 구조변이(genomic rearrangement) 분석을 통해 암을 일으키는 유전변이를 찾고 이것의 통계적 유의성, COSMIC 등에 보고된 자료등과 종합하여, 발암 유전자(driver gene)로 확인하는 과정, 단백질 비 부호화(non-coding) 영역의 유전변이 확인, 기계학습 알고리즘(NMF)을 이용한 변이 패턴(mutational signature) 분석, 특정 유전자의 유전변이와 변이 패턴, 그리고 구조변이와의 상관관계 분석에 이르기까지 다양한 분석과정을 함께 할 수 있었습니다.
이번 연구의 가장 큰 의의라면, 그동안 알지 못했던, 단백질 비 부호화 영역, 즉 유전체의 95% 영역에서 의미 있는 발암 기전이 있는지 확인했다는 것입니다. 2001년 인간유전체 프로젝트 이후, 전장유전체 해독 분석이 증가하긴 했지만 비용, 분석 등 문제로 암 유전체까지 전장유전체를 분석하기 어려웠고, 주로 단백질 부호화 영역만 서열 결정(exome sequencing)하여, 유전변이를 확인해 왔습니다. 이번 전장유전체 분석으로 의미있는 유전변이가 단백질 비 부호화 영역에 있는지, 구조적으로 유전체가 어떻게 변화하는지(Structural variation)를 확인할 수 있었습니다. 분석 결과, 일부 높은 빈도의 유전변이 좌위가 있긴 하지만, 전체적으로 큰 영향을 주는 것은 아니라고 합니다.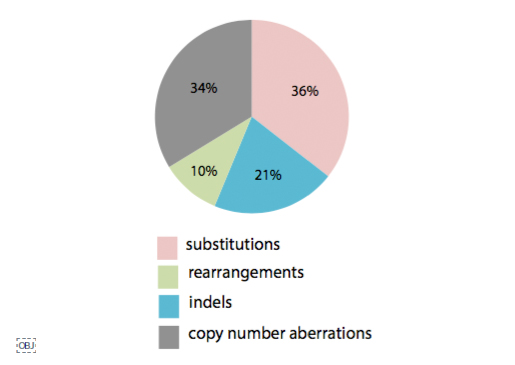
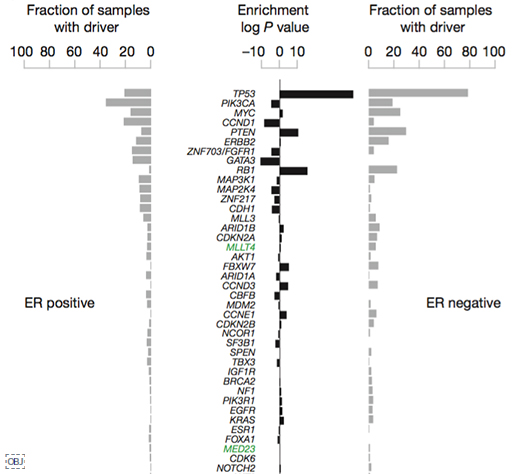
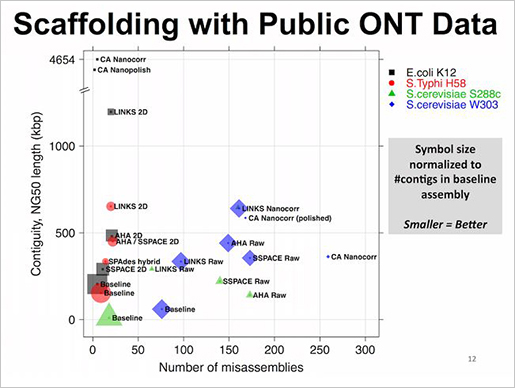
이번 연구를 통해 발암 유전자 93개에서 1,628개의 유전변이를 발견했습니다. 10개 유전자의 발암 변이(driver mutation)가 전체 발암 변이의 62%를 차지한다고 합니다. 위 그림은 이번 연구에서 발견한 발암 유전자를 에스트로겐 수용체 양성(ER+), 음성(ER-) 각각 나눴을 때 발견된 빈도를 순서대로 보여줍니다. 녹색으로 표시된 유전자 MLLT4, MED23은 이번 연구로 새롭게 발견한 발암 유전자입니다. 예전에도 많이 알려졌던 것 처럼 ER+에는 Oncogene PIK3CA 과발현과 변이가, ER-에는 Tumor suppressor gene TP53의 변이가 가장 많이 발견되었습니다.
연구팀은 치환 변이, 구조 변이의 패턴을 확인하기 위해 별도로 변이 패턴(mutational signature) 분석을 수행하였습니다. 얼굴인식에도 사용되는 기계학습 알고리즘 가운데 하나인 NMF(non-negative matrix factorization)을 이용하여, 전장유전체내 발견되는 유전 변이의 패턴을 구분하였고, 각 패턴이 특정 생물학적 원인과 관련되어 있음을 확인했습니다.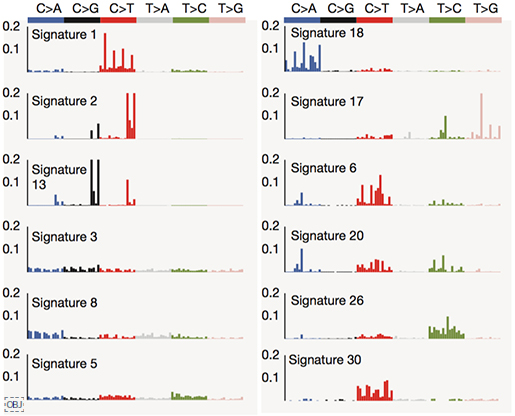
- 12개의 변이 패턴을 확인함
이 가운데, 1, 2, 8, 13번 시그니처가 이번 연구에서 유의미하게 자주 발견되었으며, 각각 생물학적인 원인과 관련되어 있습니다.
- Signature 2, 13 : APOBEC deaminase 활성과 관련됩니다. 이 효소는 바이러스의 DNA/RNA에 변화를 만들어 감염을 억제하는 역할을 하지만, 변이가 있을 경우 발암 가능성이 높아집니다.
- Signature 8 : BRCA1/2 유전자의 고장으로 정상적인 DNA 수복(DNA repair)을 못할 때 이러한 패턴의 변이가 발견됩니다. 특히 다양한 구조변이와 관련됩니다.
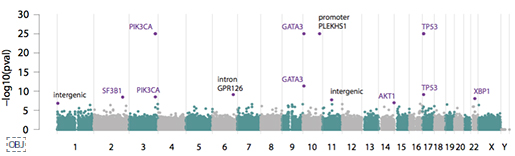
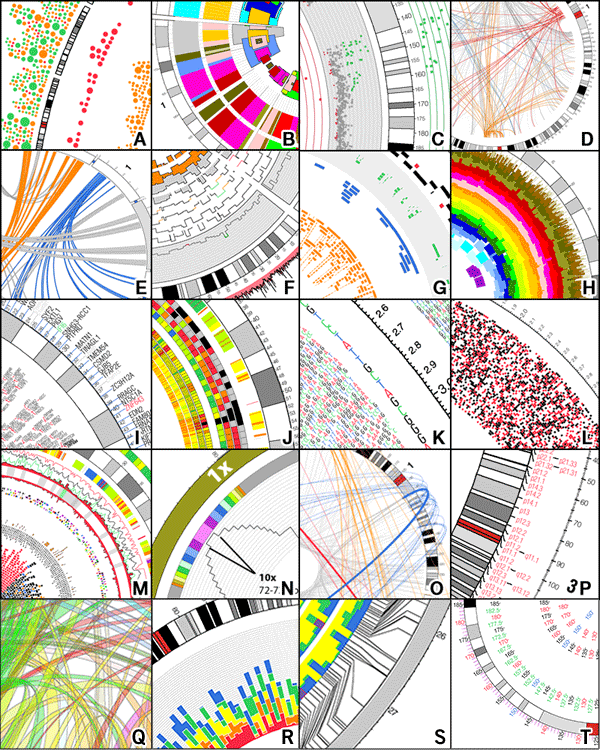
<그림4>는 전장유전체로 유의한 발암 영역을 Manhattan plot으로 확인한 결과입니다. 기존에 잘 알려져 있던 발암 유전자들과 함께, 새롭게 발견된 non-coding 영역의 변이도 함께 알 수 있습니다. 이 가운데 가장 특이했던 것은 PLEKHS1의 프로모터 영역으로, 저 영역의 변이가 있으면, Signature 2, 13번이 높아, APOBEC 효소 활성과 관련있음을 말해줍니다.
이번 연구 결과는 바로 EGA에 공개되어(EGAS00001001178) 전세계 연구자들에 의해 암 유전체를 더욱 상세히 연구하게 할 것이며, 암 맞춤의료의 기반 자료로 사용되어 인류의 암 정복을 위한 전환점이 될 것으로 기대되고 있습니다. 이러한 맞춤의료, 정밀의료의 시대에는 대규모 생물정보 데이터의 분석과 해석, 관련 데이터베이스와 지식베이스의 구축이 최고의 전문가들과 함께 이루어져야 합니다. 암 유전체의 충분한 해석과 이해, 그리고 맞춤치료에 대한 가능성은 이후, 모든 인간의 질병도 극복할 수 있게 할 것이며, 더 나아가 유전체 정보에 따라 미리 질병에 걸리지 않도록 예방할 수 있는 영역까지 확장될 것입니다.
(주)인실리코젠의 연구진은 본 연구의 노하우를 통해 앞으로 정밀의학, 맞춤의학 더 나아가 질병없는 인간의 미래에도 중요한 생물정보 가치를 만들 수 있도록 노력하겠습니다.김형용 수석개발자
Posted by 人Co
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/207






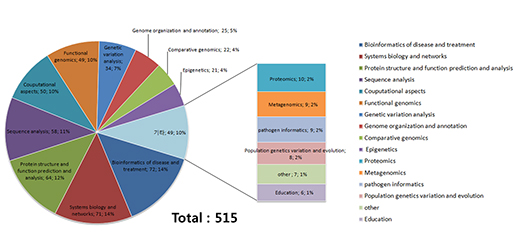
 Figure 2. Exhibitors of ISMB/ECCB2015
Figure 2. Exhibitors of ISMB/ECCB2015