약물 대사 변경으로 조절되는 잠재적인 drug-drug interaction 확인하기
- Posted at 2010/10/25 08:43
- Filed under 제품소식
Can I identify potential drug-drug interactions mediated by alterations of drug metabolism?
drug 대사의 변경에 의해 조절되는 잠재적인 drug-drug interactions를 확인 할 수 있는가? 항응고제로 혈액응고를 방지하기 위한 약물로 알려진 쿠마딘의 대사에 대해 알아보고자 한다. 쿠마딘(와파린)은 항응고를 하는 약제로 혈관안에서 혈전이 형성되는것을 막아주기 때문에 주로 혈전 및 색전증 치료에 쓰이고 있다. 쿠마딘은 주로 간에서 대사되는데, 간 대사효소인 CYP3A4에 의해 미량 대사된다고 알려져 있다. PathwayStudio를 통해 쿠마딘과 CYP3A4의 관계를 알아보고 CYP3A4에 영향을 주는 약물에 대해 조사해봄으로써 durg-drug 상호작용을 확인해보고자 한다.
Step to follow
Step 1. Coumadin 검색
Information pane에서 coumadin을 검색한다. 검색된 coumadin을 복사하고 새 pathway 문서에 붙여넣기를 한다.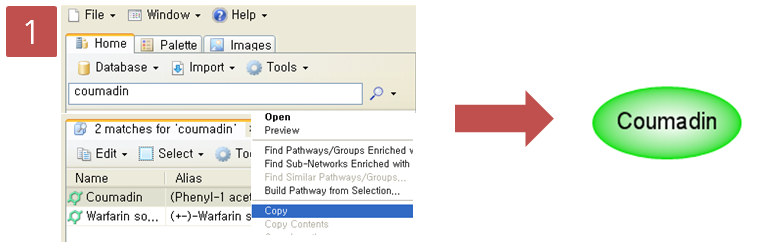
Step 2. Pathway 옵션 설정 및 Pathway 확인
coumadin이 어떤 효소에 의해 대사되는지 알아보 pathway로 나타내기 위해 옵션 설정 과정을 거친다. Advanced Build Pathway Wizard 에서 Add Neighbors > Directionality: “upstream” > Entity type: “protein” > Filter Parameters: “ChemicalReaction” 순으로 선택한다.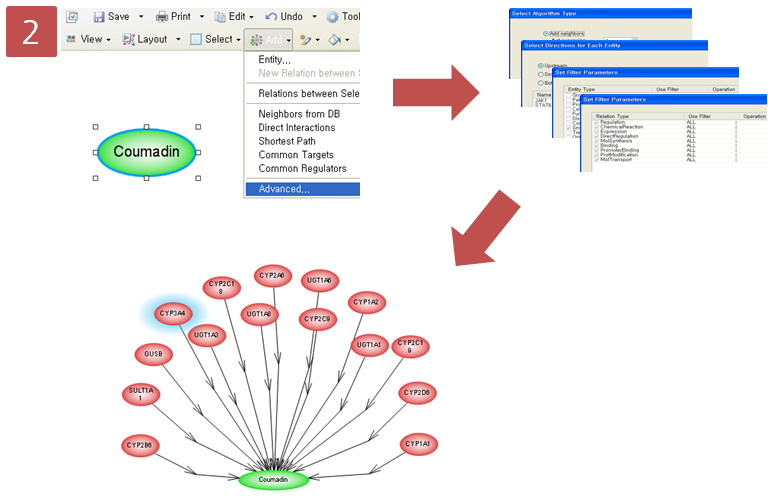
Step 3. Pathway 옵션 설정
coumadin의 대사에 관여하는 효소 15개를 확인하였고, 그 중에서 CYP3A4라는 효소는 다시 어떤 small molecule에 의해 영향을 받는지 알아보기 위해 pathway 찾기를 재수행한다. CYP3A4를 선택하고 Advanced Build Pathway Wizard 에서 Add Neighbors > Directionality: “upstream” > Entity type: “small molecule” > Filter Parameters: “DirectRegulation” 순으로 선택한다.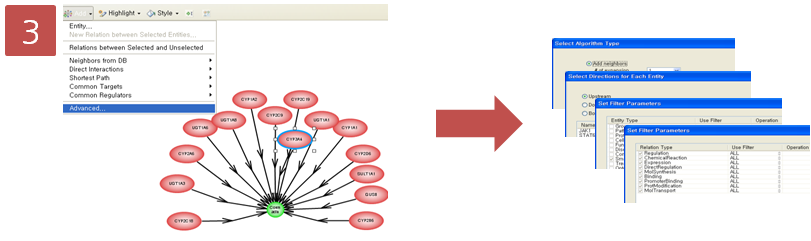
Step 4. Active Style 변경
Effect와 Reference 개수에 따라 그래프 보기에서도 효과를 나타내 줄 수 있다. Style 메뉴의 Active Style Sheet에서 By Effect를 선택하면 Effect의 Positive, Negative 효과에 따라 Relation 선색을 다르게 할 수 있으며, By Reference Count를 선택하면 Reference의 개수에 따라서 Relation 선색이 달라지는 것을 확인 할 수 있다.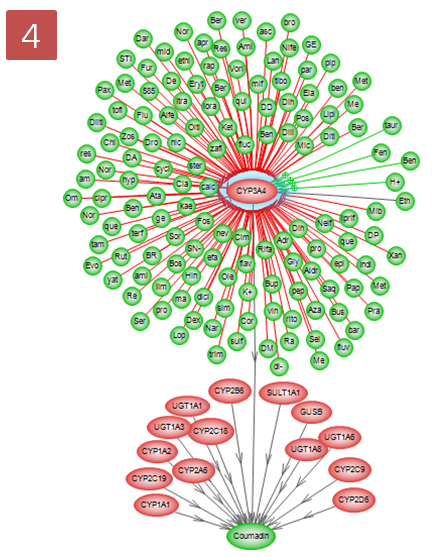
아래 동영상보기를 하시면 4개의 Step을 한 번에 보실 수 있습니다.
Posted by 人Co
- Tag
- ChemicalReaction, Coumadin, CYP3A4, Drug, insilicogen, interaction, metabolism, pathway, protein, Reference, upstream, 대사효소, 인실리코젠, 쿠마딘
- Response
- No Trackback , No Comment
- RSS :
- https://post-blog.insilicogen.com/blog/rss/response/85